DOI:
10.1039/C1SC00154J
(Edge Article)
Chem. Sci., 2011,
2, 1554-1559
Group 13 BN dehydrocoupling reagents, similar to transition metal catalysts but with unique reactivity†
Received
16th March 2011
, Accepted 28th April 2011
First published on 2nd June 2011
Abstract
Reactions of the AlIII and GaIII bases Al(NiPr2)3 and E(NMe2)3 (E = Al, Ga) with the amine-boranes [iPr2NHBH3] and [tBuNH2BH3] give the amino-borane monomer [iPr2N = BH2] (4) and the borazine [tBuNBH]3 (5), respectively. This is similar to the results of dehydrocoupling previously seen with single-site RhI catalysts and appears to occur via intermediate group 13 hydrides, as shown by the isolation of the amido-alane [H2Al(μ-NiPr2)]2 (7) in the formation of 4 from Al(NiPr2)3. In general, the outcome of group 13 dehydrocoupling reactions show a marked dependence on the amine-borane used and on the nucleophilic and redox character of the group 13 pre-catalyst. The importance of these factors is seen in the formation of the unusual, delocalised amino-borane [B{(NHBH)N(SiMe3)Si(Me2)N(SiMe3)2}3] (10) in the non-catalytic reaction of [Ga{N(SiMe3)2}3] with [NH3BH3], in which coupling of B–N as well as Si–N bonds occurs along with the deposition of Ga metal.
Introduction
The pre-eminence of transition metals in homogeneous and heterogeneous catalysis can be traced to the possession of valence d-orbitals and to the unique activation of metal-bonded ligands arising from synergic bonding.1 Additional important features that give particularly high activity are rapid ligand and redox kinetics, which in particular result in energetically-accessible oxidative addition and reductive elimination reactions. It has been pointed out recently, however, that many of the factors that are responsible for high catalytic activity in transition metals can also be found elsewhere in the periodic table, despite the absence of accessible valence d-orbitals for bonding, and it is becoming clear that a range of main group metals might also form the basis for a broad range of single-site homogeneous catalysts.2
In the past twenty years there has been widespread interest in the applications of transition metal organometallics in a broad range of element–element bond-forming reactions via homo- and hetero-dehydrocoupling of element-H bonds (eqn 1).3,4 This type of reaction has broad applications in molecular and materials synthesis. An emerging area of interest is the use of amine-boranes, R2NHBH3, as light-weight materials for the chemical storage of H2 (eqn 2).5,6 One of the prerequisites for the applications of these species (e.g., in fuel cells) is the ability to release H2 rapidly. Hence, a great deal of effort has gone into the development of transition metal catalysts for this dehydrogenation step. Many of these species are based on precious metals, however, such as Rh, Ru and Pd.5
| | | E–H + H–E′ → E–E′ + H–H | (eqn 1) |
| |  | (eqn 2) |
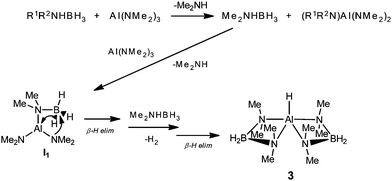
Our interest in this area has stemmed from our recent observation that the Sn(IV) metallocene Cp*2SnCl2 catalytically dehydrocouples primary phosphines in a similar manner to Zr(IV) catalysts.7,8 More recently we found that Al(NMe2)3 catalytically couples Me2NHBH3 into the ring dimer [Me2NBH2]2 (1) and the chain [(Me2N)2BH] (2) (Scheme 1).9 This reaction appears to occur through the hydride catalyst [{(Me2N)2BH2}2AlH] (3), which is generated in situ. This outcome and the intermediacy of metal hydrides is similar to that of the small number of other d0 CaII-, YIII-, and ScIII-catalysed reactions with Me2NHBH3 introduced in recent years by the groups of Harder and Hill.8 However, it is still unclear what the synthetic scope of d0 catalysis is in relation to transition metal systems on the basis of all of the studies reported so far, because of the very limited number of amine-borane precursors tested and the lack of kinetic studies.10,11 Here we show (i) that the products of reactions of a broader range of amine-boranes involving AlIII and GaIII amide pre-catalysts are similar to those formed by transition metal catalysts, (ii) that the activity of these main group catalysed reactions are comparable to some transition metal counterparts, and (iii) that the increased redox activity of GaIII bases can radically affect the course of the coupling reactions in unique ways not found in transition metal chemistry.
 |
| | Scheme 1 Formation of 1 and 2 by dehydrocoupling with the presumed catalytic intermediate 3. | |
Results and discussion
Preliminary studies of the reactions of Al(NMe2)3 with [iPr2NHBH3] (A) and [tBuNH2BH3] (B)—the problem of ligand exchange
The first series of reactions we explored were those involving Al(NMe2)3 with [iPr2NHBH3] (A) and [tBuNH2BH3] (B). In situ11B NMR experiments at room temperature showed that catalytic dehydrocoupling occurs in both of these systems to give the monomer [iPr2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2] (4) (t., δ 35.2, J11B–H = 125 Hz)12 in the case of A and the borazine [tBuNBH]3 (5) (d., δ 27.2, J11B–H = 129 Hz)5 in the case of B (Fig. 1) (ESI†). However, these reactions are complicated by initial ligand exchange of Me2N for iPr2NH and tBuNH2, leading to the formation of the previously observed hydride [{(Me2N)2BH2}2AlH] (3) (Scheme 2).9 As a result, the ring [Me2NBH2]2 (1) is also formed as a contaminant in both reactions (Scheme 1). Further evidence of the ligand exchange reaction was obtained by the isolation of the exchange product [(Me2N)2Al(μ-NHtBu)]2 (6) from the 1
BH2] (4) (t., δ 35.2, J11B–H = 125 Hz)12 in the case of A and the borazine [tBuNBH]3 (5) (d., δ 27.2, J11B–H = 129 Hz)5 in the case of B (Fig. 1) (ESI†). However, these reactions are complicated by initial ligand exchange of Me2N for iPr2NH and tBuNH2, leading to the formation of the previously observed hydride [{(Me2N)2BH2}2AlH] (3) (Scheme 2).9 As a result, the ring [Me2NBH2]2 (1) is also formed as a contaminant in both reactions (Scheme 1). Further evidence of the ligand exchange reaction was obtained by the isolation of the exchange product [(Me2N)2Al(μ-NHtBu)]2 (6) from the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction mixture of Al(NMe2)3 and [tBuNH2BH3] (A) (in 27% yield†) and its structural characterisation (Fig. 2).‡
:1 reaction mixture of Al(NMe2)3 and [tBuNH2BH3] (A) (in 27% yield†) and its structural characterisation (Fig. 2).‡
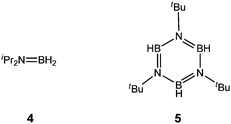 |
| | Fig. 1 Structural formulae of 4 and 5. | |
 |
| | Scheme 2 Precursor A, R1 = R2 = iPr; precursor B, R1 = tBu, R2 = H. | |
 |
| | Fig. 2 Structure of the ligand-exchange product 6. H-atoms (except those on N) have been removed for clarity. Selected bond lengths (Å) and angles (°): Al(1)–N(1) 1.9570(9), Al(1)–N(2) 1.8032(8), Al(1)–N(3) 1.8015(9), Al(1)–N(1) 1.9608(8), N(1)–Al(1)–N(1) 86.00(4), Al(1)–N(1)–Al(1) 94.00(4). | |
Reaction of Al(NiPr2)3 with [iPr2NHBH3] (A) – the potential catalytic species and the mechanism of reaction
Under these circumstances it was unclear whether 3 or an unseen hydride intermediate is the major catalytic species in dehydrocoupling to 4 and 5. However, repeating the dehydrocoupling reactions of A with Al(NiPr2)3 avoided the formation of 3 and led to the exclusive formation of the monomer [iPr2NBH2] (4) by in situ11B NMR studies. This reaction is again catalytic, with complete conversion of A to 4 in 2 h at 60 °C using a 10 mol% loading of Al(NiPr2)3 in benzene (ESI†). Interestingly, there is a relatively long induction period for this reaction before the evolution of H2 gas is observed. This is particularly noticeable with lower catalyst loadings. For example, for a 2 mol% catalyst loading of Al(NiPr2)3 to A in toluene the evolution of H2 is only observed after 20–30 mins at 20 °C. Using a 2 mol% loading at 20 °C in toluene, a TON50 of ca. 25 and a TOF of ca. 2.5 h−1 were calculated using in situ11B NMR spectroscopy. This can be compared to the higher activity found for [Rh(1,5-cod)(μ-Cl)]2 which converts A to 4 in 49% yield at 25 °C in 24 h using a 1 mol% catalyst loading in toluene (estimated TON ca. 50, TOF 2 h−1).12b
In situ
11B NMR spectroscopic studies also allowed us to investigate the kinetics of the reaction of Al(NiPr2)3 with A. A plot of ln(I/Io) (where I is the integral of A at a given time and I0 is the integral at the start of reaction) for a 2 mol% loading of Al(NiPr2)3 to A in toluene shows that the reaction is first order in A (Fig. 3).
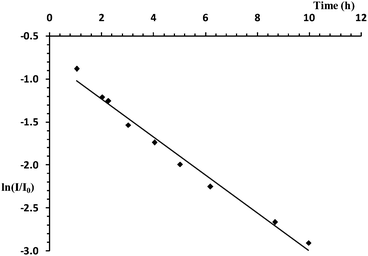 |
| | Fig. 3 Plot of time (h) vs. ln(I/I0) for the reaction of a 2 mol% loading of Al(NiPr2)3 with A in toluene (20 °C). | |
Prolonged storage of a 2:1 reaction mixture of A with Al(NiPr2)3 in toluene at −20 °C gave a small crop of crystals of the AlIII hydride [H2Al(μ-NiPr2)]2 (7).13 This was spectroscopically† and structurally characterised.‡ In the solid state the two AlIII centres of the dimeric structure of 7 are bridged by two iPr2N groups with two terminal hydrides per metal atom (Fig. 4). Crystalline 7 was subsequently shown to be an active catalyst for the dehydrocoupling of A to 4, with a ca. 50% conversion of A to 4 in 4 days using a 0.5 mol% loading of 7 at 20 °C in benzene (corresponding to TON50 of ca. 90 and a TOF of ca. 1 h−1) (ESI†). Interestingly, two minor BH4− resonances are also present at the end of this reaction [quint., δ −34.8 (J11B–H = 85 Hz), −37.0 (J11B–H = 86 Hz)]. The activity of 7 can be compared to that of [Rh(1,5-cod)(μ-Cl)]2 which converts A to 4 in 49% yield at 25 °C in 24 h using a 1 mol% catalyst loading in toluene (estimated TON ca. 50, TOF 2 h−1),12b and Cp2Ti which gives a quantitative yield of 4 in 1 h at 20 °C using a 2 mol% loading in toluene.14
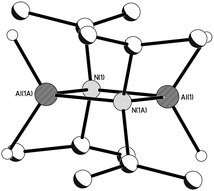 |
| | Fig. 4 The potential catalytic intermediate 7. H-atoms have been removed for clarity, except the hydrides: Selected bond lengths (Å) and angles (°): Al(1)–N(1) 1.9636(8), Al(1)–N(1A) 1.9679(8), Al(1)–H mean 1.55, N(1)–Al(1)–N(1A) 91.22(3), Al(1)–N(1)–Al(1A) 88.78(3). | |
An in situ27Al NMR spectrum of the catalytic reaction of A with Al(NiPr2)3 (10 mol%, 20 °C) shows the same single, broad resonance as that found for isolated 7 (δ 140.0).§ Thus the alane dimer 7 is at least a plausible catalyst in the formation of 4, by the tentative mechanism shown in Scheme 3. The first part of this cycle, by which 7 is generated in situ, gives the product 4 stoichiometrically. This is followed by a simple cycle involving deprotonation of [iPr2NHBH3] (A) and β-hydride elimination to regenerate 7. We attribute the presence of the BH4− anion at the end of this reaction to a potential deactivating step involving the formation of I (Scheme 3), viahydride transfer to the B atom. The observation of two BH4− resonances in the in situNMR spectroscopic study of the reaction presumably arises from ion-paired and -separated BH4− in this species. The proposed termination of this reaction is closely related to that reported in the deactivation of the dihydride pincer catalyst [(POCOP)IrH2] [POCOP = η1-1,3-(OPtBu2)2C6H3] with BH3 in its dehydrogenation reaction with ammonia borane, NH3BH3.15 The mechanism shown in Scheme 3 is consistent with the long induction period observed (especially for lower catalyst loadings), which is presumably associated with the stoichiometric formation of a significant amount of 7 prior to establishing the catalytic reaction. The observed first-order kinetics in A once the catalytic reaction takes over is also consistent with a simple catalytic cycle involving a species like 7, in which deprotonation of A and subsequent β-H elimination are rate-determining (as shown in Scheme 3).
 |
| | Scheme 3 Proposed catalytic cycle involved in the formation of 4. | |
Potential relationship to the previously reported catalytic reaction of Al(NMe2)3 with Me2NHBH3
As noted in the introduction, our previously reported study of the catalytic reaction of Al(NMe2)3 with Me2NHBH3 gives [Me2NBH2]2 (1) as the major product together with the chain [(Me2N)2BH] (2) (Scheme 1). Although the activity of Al(NMe2)3 is relatively low at room temperature, e.g., using an 8 mol% loading in toluene gives ca. 80% conversion after 5 days (TON50 = 6, TOF = 0.13 h−1), the reaction is significantly faster at elevated temperatures, e.g., a 5 mol% loading of Al(NMe2)3 at 50 °C in toluene gives complete reaction after 48 h. The reactivity at higher temperature is at the lower end of that observed for a range of transition metal catalysed systems.5a One intriguing feature of this reaction is the fact that the minor product 2 is only formed in the initial stage of this reaction and that its concentration remains static throughout the course of catalytic reactions at a ratio of ca. 1:1 with the presumed catalyst [{(Me2N)2BH2}2AlH] (3). In the light of the activity of the less sterically-encumbered dihydride 7 in the catalytic dehydrogenation of [iPr2NHBH3] (A) described in the previous section, we wondered whether a similar dihydride might in fact be involved in the reaction of Al(NMe2)3 with Me2NHBH3. Such a dihydride could be generated by β-H transfer from B to Al in 3 (Scheme 4). DFT (B3LYP/cc-pVDZ) calculations of this dissociation reaction indeed show that it is favourable by ΔG = −14.4 kcal mol−1 at 298 K (ΔG‡ = +18.7 kcal mol−1). It therefore appears likely that the dihydride 8 (Scheme 4), which is closely related to 7, is at least another potential catalytic species in this system. This dissociation will become more favourable at higher temperatures because of the entropy involved (i.e., favourable TΔS term).
 |
| | Scheme 4 DFT calculated pathway to 2 and the potential catalyticdihydride 8. | |
A plot of time vs. ln(I/I0) using our previously reported in situ11B NMR spectroscopic data on the reaction of Al(NMe2)3 (8 mol% in toluene) with Me2NHBH3 (see Figure S3, ESI ref 9†) shows that this reaction is first-order in the amine-borane Me2NHBH3 (Fig. 5). Bearing in mind the now complicated picture of this reaction (i.e., the potential involvement of the hydrides 3 and/or 8) it is difficult to find a definitive explanation for this observation. However, one plausible explanation is that like the dehydrogenation of [iPr2NHBH3] with Al(NiPr2)3 (A) (Scheme 3) deprotonation of Me2NHBH3 followed by β-hydride elimination to give Me2NBH2 is rate determining. This is consistent with the fact that Me2NBH2 is never observed in in situ11B NMR spectroscopic studies of the reaction of Al(NMe2)3 with Me2NHBH3, irrespective of the stoichiometry, presumably because it is rapidly consumed in the formation of the dimer [Me2NBH2]2 (1) in subsequent steps.9 It should be noted, however, that this situation is very different to that recently reported by Hill and coworkers in the reactions of Group 2 silylamides M{N(SiMe3)2}2 with Me2NHBH3 in which β-H elimination to Me2NBH2 appears to be faster than any other process.11
 |
| | Fig. 5 Plot of time (h) vs. ln(I/I0) for the reaction of a 8 mol% loading of Al(NMe2)3 with Me2NHBH3 in toluene (20 °C). | |
Further investigation of the catalytic reaction of Al(NMe2)3 with [tBuNH2BH3] (B)—the likely mechanism of reaction
An in situ11B NMR spectroscopic study of the reaction of [tBuNH2BH3] (B) with Al(NMe2)3 to the borazine [tBuNBH]3 (5) under catalytic conditions [3 mol% Al(NMe2)3] in benzene also suggests that an active hydride species other than 3 (or 8) is involved. No resonances which could be attributed to the hydride 3 or the chain product 2 are observed throughout the study, instead slow build up of the borazane [tBuNHBH2]3 (9) at δ −5.1 (t. J11B–H = 106 Hz) occurs after ca. 1 day at 20 °C together with the formation of an appreciable amount of the borazine 5. After 4 days at 20 °C 30% of B has been converted into products; 9 (8%), 5 (13%) and an unidentified (probably polymeric) species at δ 30.9 (8%).16 The apparent intermediacy of the borazane 9 in the reaction of B with Al(NMe2)3 is similar to the dehydrocoupling reactions of a range of amine-boranes [RNH2BH3] (R = H, Me, Ph) with the transition metal catalyst [Rh(1,5-cod)(μ-Cl)]2 which also involve the formation of borazanes, which are dehydrogenated in a second step to the borazines.12b However, in the case of the RhI-catalysed reactions the borazanes are formed cleanly in 4–6 h followed by a second step in which the borazines are then formed over a period of 48–72 h. Thus, the rate of formation of the borazanes is much faster than that of the subsequent dehydrogenation to the borazines in this case. Assuming that the borazane 9 is the intermediate in the formation of the borazine 5, our results suggest that the rate of production of 9 is slower than its subsequent dehydrogenation to 5 using Al(NMe2)3 as the pre-catalyst. The persistence of the borazane 9 which (unlike the RhI-catalysed reaction) is present at the end of the reaction may be associated with the lifetime of the catalytic intermediate(s) involved in the dehydrogenation of 9 to 5. Indeed, the lower redox stability of Ga(NMe2)3 (and of the likely GaIII hydride intermediates) leads to a much slower reaction with B than with Al(NMe2)3 and to major decomposition into Ga metal after 4 days at 20 °C. Significantly, in this case more of 9 is present in the reaction after this time than at the end of the reaction involving Al(NMe2)3 (see ESI†).
Storage of the 2:1 stoichiometric reactions of B with E(NMe2)3 (E = Al, Ga) in toluene at −20 °C gives small amounts of the crystalline borazane [tBuNHBH2]3 (9).§ Higher yields were obtained for Ga(NMe2)3 experiments compared to the reaction involving Al(NMe2)3, consistent with in situ11B NMR spectroscopic studies (ESI†). Crystals of 9 were characterised by 1H and 11B NMR spectroscopy,§ as well as by a single-crystal X-ray structure.‡ Although the crystals are of relatively poor quality (with R1 = 0.124), the solid-state structure is unambiguous (Fig. 6). All four crystallographically-independent molecules found in the hexagonal unit cell have a similar twist-boat shape, in which the tBu groups are placed equatorially. DFT calculations at the B3LYP/cc-pVDZ level reproduce this conformation and the bond lengths and angles well (ESI†).
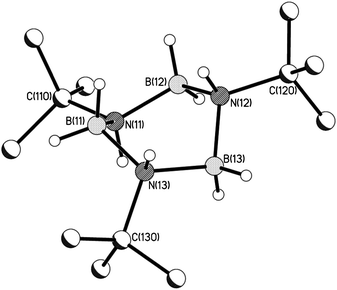 |
| | Fig. 6 Molecular structure of the borazane 9. Only one of the four crystallographically-independent molecules in the unit cell is shown. H-atoms on the tBu groups are also not shown for clarity. Selected bond lengths (Å) and angles (°) (means over all four independent molecules): B–N 1.58(1), N–B–N 108.6(8), B–N–B 111.2(8). | |
Reaction of Ga{N(SiMe3)2}3 with NH3BH3
One important observation which was made in all of the catalytic reactions involving Ga(NMe2)3 is the formation of Ga metal. It is well known that GaIIIhydrides, the likely catalytic intermediates in these reactions, are thermally unstable and this is likely to limit the lifetimes of catalysts based on this metal. We have seen recently how reductive elimination can limit the lifetimes of SnIV dehydrocoupling catalysts involving P–P bond formation.5 What was less clear is how the reduction of GaIII to Ga metal can radically alter the reaction pathway observed. A dramatic example of this is seen in the current study when we explored the dehydrocoupling of ammonia-borane NH3BH3 (C) with group 13 amides.17 The unexpected product of the stoichiometric reaction of Ga{N(SiMe3)2}3 with C is [B{(NHBH)N(SiMe3)Si(Me2)N(SiMe3)2}3] (10), in which a combination of B–N and Si–N bond formation has occurred (Fig. 7). Owing to the thermal instability of 10, which decomposes steadily during the course of ca. 1 h at room temperature into a grey solid (with the apparent liberation of hydrogen), we were unable to obtain satisfactory analytical data. However, the room-temperature 11B NMR spectrum shows the presence of a quartet at δ −24.2 (q., 2J11B–H = 96 Hz) for the central B atom [B(0)] together with a broad doublet at ca. δ +30 for the other boron environments [B(11,21,31)]. The presence of an additional quintet at δ −43.7 appears to arise from the decomposition of 10 into BH4−. The 29Si NMR shows the expected three resonances for the N(SiMe3), SiMe2 and N(SiMe3)2 groups found in the solid-state structure.
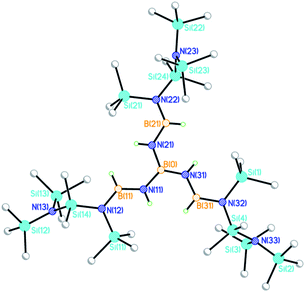 |
| | Fig. 7 Molecular structure of 10. Disorder of one of the N(SiMe3)Si(Me2)N(SiMe3)2 groups has been omitted for clarity. Selected bond lengths (Å) and angles (°); B(0)–N(11,21,31) range 1.37(2)–1.46(2), N(11,21,31)–B(11,21,31) range 1.40(2)–1.47(2), B(11,21,31)–N(12,22,32) range 1.41(2)–1.49(2), Si–N range 1.64(2)–2.03(2), N–B(0)–N range 118(1)–121(1), B4N6 core atoms within 0.1 of a plane. | |
The molecular structure of 10 consists of a B(NBN)3 core arrangement which is built around a central trigonal-planar boron atom [B(0)] (Fig. 7).‡ Each of the three arms of the structure is terminated by an N(SiMe3)Si(Me2)N(SiMe3)2 group (Fig. 7). The near planarity of the entire B(NBNsilyl)3 subunit of 10 (with all ten atoms within 0.1 Å of a plane) suggests extensive delocalisation throughout this portion of the molecule viapπ–pπ B⋯N bonding. This structural and bonding arrangement is similar to that of [B{N(H)BMes2}3] (Mes = 2,4,6-Me3-C6H2) reported previously by Nöth and coworkers, obtained by a different route.18 The mechanism by which the unusual N(SiMe3)Si(Me2)N(SiMe3)2 ligand of 10 is generated is unclear, but formally involves the process 2(Me3Si)2N− → [N(SiMe3)Si(Me2)N(SiMe3)2]− + Me−. One clue to its origin is provided by previous reports of N–SiMe3 bond cleavage in the reactions of metal silylamide complexes with reducing agents, for example, reduction of the [Y(LPh,Ph)2] [LPh,Ph = (Me3Si)NC(Ph)CHC(Ph)N(SiMe3)] with Y metal in THF gives a complex containing a metal-bonded –NSiMe2CH2– fragment.19 The ligand fragmentation and rearrangement observed in 10 could possibly be the result of reductive cleavage by finely-divided Ga metal, generated during the reaction. However, it can be noted also that Si–C bond cleavage of the (Me3Si)2N ligand can occur in the pyrolysis reactions of GaIII silylamide complexes themselves.20
Conclusions
Studies of the reactions of AlIII and GaIII amide bases with amine-boranes show a strikingly similar pattern to transition metal catalysis both in terms of the products formed and (in some cases, like 7) in terms of activity. Like transition metal and early main group metal BN dehydrocoupling systems, the active catalytic components are metal hydrides. In the case of GaIII (whose hydrides are unstable) this results in catalyst instability and in deactivation by the formation of Ga metal. However, this undesirable behaviour can in itself lead to unusual stoichiometric side reactions, as seen in the combination of B–N and Si–N bond formation in the case of 9. Perhaps the most important conclusion from the current work, that will inform future directions in this area, is that the choice of p-block metal is key to redox stability in these systems and that this can be used to modulate catalytic and/or stoichiometric behaviour and the nature of products formed.
Acknowledgements
We thank the EPSRC (DSW), The Leverhulme Trust (DSW), Cambridge University (RLM), Studienstiftung des deutschen Volkes (MMH) for financial support. We also thank Dr J. E. Davies for collecting X-ray data on 6, 7, 9 and 10, Dr M. Pernpointner (Heidelberg) for the use of computer facilities, and Dr D. Reed (Cambridge) for collecting MAS NMR data.
Notes and references
-
J. P. Collman, L. S. Hegedus, J. R. Norton, R. C. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books, U.S., 2nd revised edn, 1987 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS.
- S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482 RSC; D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 314 CrossRef; T. J. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634 CrossRef CAS; R. Waterman, Curr. Org. Chem., 2008, 12, 1322 CrossRef CAS; R. Waterman, Dalton Trans., 2009, 18 RSC.
- A further growing area of interest is the use non-metallic Lewis acids and frustrated Lewis pairs in this context. Like transition metal and early main group metal catalysts these systems are also proposed to go through hydride intermediates; see F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746 Search PubMed; A. Miller and J. E. Bercaw, Chem. Commun., 2010, 46, 1709 CrossRef CAS.
-
(a) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC;
(b) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023 CrossRef CAS.
- A further important issue in H2storage is regeneration of the unsaturated, dehydrogenated products (‘spent fuel’) back to amine-boranes, see A. A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner III, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Science, 2011, 331, 1426 Search PubMed.
- V. Naseri, R. J. Less, M. McPartlin, R. E. Mulvey and D. S. Wright, Chem. Commun., 2010, 46, 5000 RSC.
- See also: R. J. Less, R. L. Melen, V. Naseri and D. S. Wright, Chem. Commun., 2009, 4929 Search PubMed.
- H. J. Cowley, M. S. Holt, R. L. Melen, J. M. Rawson and D. S. Wright, Chem. Commun., 2011, 47, 2682 RSC.
-
(a) J. Spielmann, M. Bolteb and S. Harder, Chem. Commun., 2009, 6934 RSC;
(b) D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem.–Eur. J., 2010, 16, 8508 CrossRef CAS;
(c) M. S. Hill, G. Kociok-Köhn and T. P. Robinson, Chem. Commun., 2010, 46, 7587 RSC;
(d) J. Spielmann, D. Piesik, B. Wittkamp, G. Jansen and S. Harder, Chem. Commun., 2009, 45, 3455 Search PubMed.
- During the revision stage of this paper a further report of the reactions of group 2 silylamides was published utilising (CH2)4NHBH3 and iPr2NHBH3 as precursors. These studies also support the view that the products formed in by early main group catalysts are similar to transition metal systems. See M. S. Hill, M. Hidgson, D. J. Liprot and M. F. Mahon, Dalton Trans., 2011 10.1039/c1dt10171.
- Lit. δ 34.2, J = 123 Hz;
(a) R. K. Kanjolia, L. K. Krannich and C. L. Watkins, J. Chem. Soc., Dalton Trans., 1986, 2345 RSC;
(b) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS.
- The structure of 7 has been disclosed previously as a personal communication to the Cambridge Crystallographic Data Base, but no synthetic procedure for its formation was provided,
N. Nieger, S. Schulz, private communication, CCDC 244680.
- M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831 CrossRef CAS.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekeym and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS; T. J. Hebden, M. C. Denny, V. Pons, P. M. B. Piccoli, T. F. Koetzie, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812 CrossRef CAS.
- %'s are calculated on the basis of monomers of the products 5, 9 and the unidentified polymeric product.
- The reaction of Al(NMe2)3 with NH3BH3 is not catalytic, giving the borazine [B3N3H6] and an insoluble polymeric material, which MAS NMR shows contains B and Al and IR shows contains B–H and N–H. This is apparently not the polymer [BH2NH2]n. See A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. Schmedt auf der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332 Search PubMed.
- D. Männig, H. Nöth, H. Prigge, A.-R. Rotsch, S. Gopinathan and J. W. Wilson, J. Organomet. Chem., 1986, 310, 1 Search PubMed.
- A. G. Avent, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2004, 2272 RSC.
- B. Luo, V. G. Young Jr and W. L. Gladfelter, J. Organomet. Chem., 2002, 649, 268 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: In situ11B NMR spectra on selected reactions, DFT calculations on selected species. CCDC reference numbers 817717–817720. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00154j |
| ‡ Crystals data: Compound 6, C16H44Al2N6, M = 374.52, monoclinic, space groupP21/n Z = 4, a = 8.7047(2), b = 15.6298(3), c = 9.3874(2) Å, β = 115.9220(10)°, V = 1148.68(4) Å3, μ(Mo-Kα) = 0.137 mm−1, ρc = 1.083 Mg m−3, T = 180(2)K. Total reflections 14062, unique 3651 (Rint = 0.030). R1 = 0.036 [I > 2σ(I)] and wR2 = 0.100 (all data). Compound 7; C12H32Al2N2, M = 258.36, monoclinic, space groupC2/c, Z = 4, a = 14.6013(3), b = 15.4860(3), c = 8.6751(2) Å, β = 122.28(3)°, V = 1658.4(6) Å3, μ(Mo-Kα) = 0.158 mm−1, ρc = 1.035 Mg m−3, T = 180(2)K. Total reflections 12424, unique 2880 (Rin = 0.025). R1 = 0.038 [I > 2σ(I)] and wR2 = 0.108 (all data). Compound 9, C12H36B3N3, M = 254.87, hexagonal, space groupP6(5), Z = 24, a = b = 19.5895(6), c = 31.1522(8) Å, V = 10353.0(5) Å3, μ(Mo-Kα) = 0.055 mm−1, ρc = 0.981 Mg m−3, T = 260(2)K. Total reflections 262529, unique 41471 (Rint = 0.055). R1 = 0.124 [I > 2σ(I)] and wR2 = 0.386 (all data). H-atoms on N and B were fixed geometrically. Compound 10. THF, C37H113B4N9OSi12, M = 1080.68, hexagonal, space groupP6(3), Z = 6, a = b = 29.3467(5), c = 14.9405(4) Å, V = 11143.3(4) Å3, μ(Mo-Kα) = 0.240 mm−1, ρc = 0.966 Mg m−3, T = 180(2)K. Total reflections 20082, unique 4976 (Rint = 0.056). R1 = 0.124 [I > 2σ(I)] and wR2 = 0.334 (all data). H-atoms on N and B were fixed geometrically.Data were collected on a Nonius KappaCCD diffractometer, solved by direct methods and refined by full-matrix least squares on F2 (G. M. Sheldrick, SHELX-97, Göttingen, 1997). CCDC 817717–817720 contain the supplementary crystallographic data for 6, 7, 9 and 10. Data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/conts/retrieving.html (or from The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax 441223 316033. |
| § Synthesis of[(Me2N)2Al(μ-NHtBu)]2 (6): Al(NMe2)3 (2.3 ml of a 1 mol dm−3 solution in n-hexane) was added to a solution of [tBuNH2BH3] (200 mg, 2.3 mmol) in THF (15 ml) resulting in a colourless solution. The solution was stirred for 4 h and the solvent removed in vacuo until a saturated solution was formed. Storage of the solution at −20 °C overnight results in colourless crystals of the product (115 mg, 0.61 mmol, 27%) which decomposed above 300 °C. IR (Nujol, NaCl windows), ν/cm−1 = 2928 (s, broad, v N–H), 2306s, 2246s, 1462s, 1372m, 1264m, 1206m, 1165m, 1130m, 1045s, 980s, 938s. 832s, 791s, 741s, 610s. 1H NMR (500.2 MHz, δ (ppm), d8 – THF, +25 °C): 3.6 (s, 12H, –Me), 1.7 (s, 9H, –Me (tBu)) (the N–H protons could not be identified).Characterisation of[H2Al(μ-NiPr2)]2 (7): 1H NMR (500.2 MHz, δ (ppm), d8 – toluene, +25 °C): 3.67 (sept., 2H, J = 6.8 Hz, C–H iPr), 2.22 (s., 2H, Al–H), 1.40 (d., 12H, J = 6.8 Hz, Me iPr). 27Al {1H-coupled} NMR (130.3 MHz, δ (ppm), d8 – toluene, +25 °C, rel. to Al(NO3)3/D2O), 140.0 (s.).Synthesis of[tBuNHBH2]3 (9): Ga(NMe2)3 (500 mg, 2.5 mmol) was dissolved in toluene (10 ml) and the solution added to tBuNH2BH3 (430 mg, 4.9 mmol) in toluene (10 ml). The reaction was stirred at room temperature for 24 h and the solvent removed in vacuo until 5 ml of a clear colourless solution remained. Storage at −20 °C afforded the cyclic trimer [tBuNH2BH3] as colourless crystals (30 mg, 0.12 mmol, 7%). 1H NMR (500.2 MHz, δ (ppm), d8 – THF, +25 °C). 11B NMR (160.5 MHz, δ (ppm), d8-toluene, +25 °C, rel. to BF3·Et2O/CDCl3) δ −5.1 (t. J11B–H = 106 Hz).Synthesis of[B{(NHBH)N(SiMe3)Si(Me2)N(SiMe3)2}3] (10): Ga{N(SiMe3)2}3 (5.5 g, 10 mmol) in THF (10 ml) was added to NH3BH3 (617 mg, 20 mmol) in THF (10 ml) at room temperature. The reaction mixture was stirred for ca. 90 mins after which a mass of (unidentifed) grey-white powder was formed. Filtration through Celite to gave a clear solution. The majority of the solvent was removed until ca. 5 ml remained. Storage of the solution at −20 °C afforded colourless crystals of 9 (0.125 g, 0.12 mmol, 3%). 11B NMR (160.5 MHz, δ (ppm), d8-toluene, +25 °C, rel. to BF3·Et2O/CDCl3), ca. + 30 (br. d. obscured by resonance due to borosilicate of NMR tube), −24.2 (q., 2J11B–H = 96 Hz), also present −43.7 (quint., J11B–H = 82 Hz, BH4−). 29Si NMR (99.4 MHz, δ (ppm), d8-toluene, +25 °C, rel. to Me4Si), 1.02 (SiMe3), 2.26 (SiMe3), −24.0 (SiMe2). |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.