Alkynes as an eco-compatible “on-call” functionality orthogonal to biological conditions in water
Nick
Uhlig
and
Chao-Jun
Li
*
Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, Québec H3A 2K6, Canada. E-mail: cj.li@mcgill.ca; Fax: +(514)-398-3797; Tel: +(514)-398-8457
First published on 5th May 2011
Abstract
This mini review summarizes some recent examples of the use of alkyne functionality as a latent reactive group under biologically compatible conditions. Such reactions provide potential chemical tools for a wide range of applications in biological systems.
Introduction
Recent advances in chemical biology and synthetic biology desire chemical and synthetic methods with high orthogonality to the functionality of biological molecules. That is, the use of reaction partners with highly focused reactivity that will not participate in side reactions with the vast array of functionalities present in biological systems. Functional groups that are inert towards various functionalities existing under biological conditions, such as amines, acids, water, esters, thiols, and hydroxyls, yet can be chemically “turned on” with molecular level precision by some specific controlling element are highly attractive in this respect.The marriage of high orthogonality with useful reactivity can be restrictive, but alkynes, being relatively stable and unreactive toward various biological elements yet becoming highly reactive with use of an appropriate “trigger”, provide some interesting opportunities. Such triggering can occur with an alkyne or its reaction partner by using certain triggers, such as a trace amount of metal catalyst, a light source, a thermal process, molecular strain, a reactive intermediate generated in situ or a simply hydrophobic pocket in water. Reactivity wise, alkynes possess two general reaction modes: the reaction of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds (mode a) and the reaction of C–H bonds (mode b, for terminal alkynes) (Fig. 1). Both alkyne reactivities are relatively insignificant under biological conditions in the absence of a triggering element, such as a catalyst. Recently, various methods have been developed to trigger both reaction modes under biological conditions in water. This mini-review highlights some examples on this subject.
C bonds (mode a) and the reaction of C–H bonds (mode b, for terminal alkynes) (Fig. 1). Both alkyne reactivities are relatively insignificant under biological conditions in the absence of a triggering element, such as a catalyst. Recently, various methods have been developed to trigger both reaction modes under biological conditions in water. This mini-review highlights some examples on this subject.
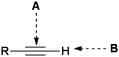 | ||
| Fig. 1 The two general “on-call” reactivity modes of an alkyne. | ||
Mode A: Reaction of the alkyne C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C bond
C bond
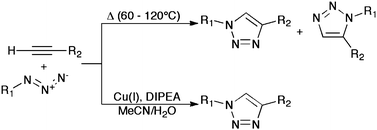
The most widely recognized bioorthogonal “on-call” reaction taking advantage of the alkyne CC bond is its use in the Huisgen 1,3-dipolar cycloaddition with azides. This reaction is one of several that has seen widespread use since its inclusion in the paradigm of “click chemistry”, as set out by K.B. Sharpless in 2001.1 The reaction is most often seen as an aqueous/organic process catalysed by copper(I) salts, the use of which affords both a dramatic rate enhancement and an increase in regioselectivity by virtue of a stepwise mechanism,2 known colloquially as the copper azide–alkyne click reaction (CuAAC). The CuAAC (Scheme 1) has become something of a poster child for the field of click chemistry, with good reason. Azide and alkynegroups are facile to introduce, have limited reactivity and possess excellent kinetic stability. Finally, the product of this reaction, a 1,2,3-triazole, is stable under biological conditions and is difficult to oxidize or reduce.3 These are ideal characteristics for a chemical tool in biological systems.
Click modification of DNA and oligonucleotides
There is perhaps no greater indication of the CuAAC's bioorthogonality than its use in the modification of DNA. Since 2006, the field of nucleotide and DNA modification using click chemistry has expanded rapidly.4 Generally, the methods used for DNA modification are similar to the original procedures, with the added necessity of a copper binding ligand, such as tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), in order to slow or prevent copper-induced DNA damage or degradation of the catalyst itself.5 One of the first groups to investigate this application of click chemistry was that of Carrell, with their seminal work on the labelling6 and metallization7 of DNA using CuAAC methods. Carell's initial work on the use of alkynyl-modified nucleobases also yielded the first example of CuAAC modification of PCR products with azidogalactose at pre-alkynyl-modified cytidine residues, in this case of up to 2000 bp in length, with no signs of DNA degradation.6 This work was furthered by the development of labeling strategies applicable to alkynyl-dT, -dA, and -dG residues as well.8 Further work on the PCR incorporation of a variety of alkynyl- and azido-modified nucleotides revealed generally excellent tolerance of alkynylgroups of different lengths, but poor acceptance of an azido-functionalized thymine derivative.9The post-synthetic modification and labeling of DNA products has progressed significantly since the initial work done by Carell. Seela and coworkers investigated the stability of alkyne functionalities of different lengths in solid-phase-synthesized oligonucleotides, as well as their labelling with coumarin fluorescent tags. The stability of base pairs was found to be improved by the presence of 1,7-octadiynyl modifications,10 and upon labeling of oligonucleotides with hydroxycoumarin, found nucleobase-specific fluorescence quenching, useful for measuring conformational dynamics in solution.11 Seela's use of tripropargyl amine functional groups also allowed bis-functionalization of oligonucleotides using both hydroxycoumarins and AZT.12
Multiple successive ligations of up to three different reporter groups to alkyne-functionalized DNA were accomplished recently by Carell and coworkers (Scheme 2), using different protection strategies.13 By taking advantage of the differing cleavage rates of TMS and TIPSprotecting groups, they accomplished the ligation of a diverse set of sensitive reporter molecules, including biotinprobes, coumarin fluorophores, azidogalactose, and azo-dyes, among others, in various combinations. The labeling was shown to be effective when the alkynyl unit was incorporated onto the nucleobase, or onto the phosphate moiety.
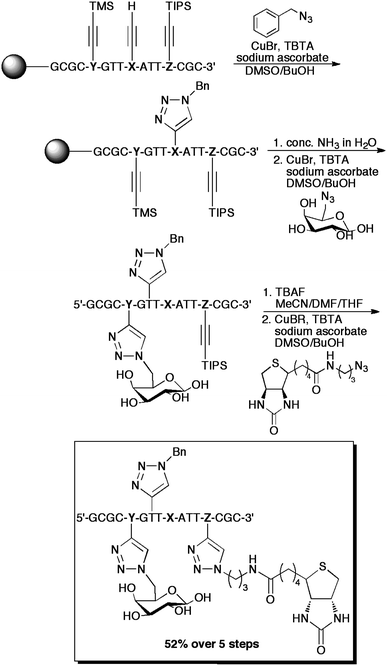 | ||
| Scheme 2 An example of the “click-click-click” procedures employed by Carell et al. for the triple-labelling of DNA. NucleotidesX, Y, and Z correspond to 5-octadiynyluracil, TMS-protected 5-octadiynylcytosine, and TIPS-protected 5-octadiynylcytosine, respectively. Conveniently, the cleavage of the oligonucleotide from the resin and the removal of the TMSgroup occurred concurrently. | ||
Recently, Brown and Graham accomplished the ligation of an HIV-derived cell penetration peptide (Tat) to oligonucleotides using CuAAC.14 Curiously, the ligation only occurred when the alkyne moiety was located on the peptide, and the azide functionality was at the 3′ end of the oligonucleotide. Previously, conjugates of peptides with oligonucleotides and peptide nucleic acids had been accomplished by Kumar and coworkers.15 Other functional biomolecules have been successfully conjugated to nucleotides as well. Inhibition of hepatitis C virus translation was accomplished by Barthélemy and coworkers by conjugating lipid moieties to oligonucleotides.16 These oligonucleotides represented antisense strands that were complementary to subdomain IIId of the internal ribosomal entry site (IRES) of hepatitis C, and the lipid conjugates thereof increased cell penetration, yielding IC50 values of 1–2 μM in cultured Huh7 cells, effectively suppressing HCVtranslation.
Seela and Brown have both accomplished the ligation of multiple DNA strands using CuAAC. In 2007, Brown utilized intramolecular click reactions to form circular single-stranded DNA, which was capable of acting as a template for a second strand to perform an intramolecular click reaction, forming pseudohexagonal double-stranded DNA catenates.17 Brown also accomplished the ligation of complementary DNA strands, utilizing base pairing as a template for the reaction.18 Strategic incorporation of alkyne- and azide-bearing uracil derivatives with varying linker lengths by phosphoramidite synthesis allowed complementary strands to be ligated using click reactions. In all cases, the duplexes saw an increase in fluorescence and ultraviolet melting temperatures, especially when the triazolyl linker was formed in an A–T rich region of the oligonucleotides.
Seela utilized single-stranded DNA with alkyne-modified 8-aza-7-deazapurine-2′-deoxyribonucleosides, related to dA and dG, with bivalent azides to produce identical cross-linked single-stranded DNA, which was then hybridized with complementary strands to produce “four-stranded DNA”.19 The click ligation could also be adapted to affect only a single click of the bivalent azide, indicating that non-identical strands could be cross-linked in a stepwise manner.
Matsuda et al. also reported the construction of double-stranded dumbbell oligodeoxynucleotides for use as decoy molecules.20 By annealing complementary 11-mer strands with 5′-azido and 3′-alkynyl functionalities and cross-linking the termini using CuAAC, they created highly stable dumbbell oligonucleotides with varying loop lengths. The dumbbell molecules were found to bind effectively to nuclear factor κB. Dumbbell oligonucleotides have already shown potential as therapeutic agents for down-regulating transactivation,21 and the use of click chemistry significantly simplifies their synthesis and allows greater tunability in their loop length, which was shown to be an important factor in binding.
Modifications of peptides, peptidomimetics, and proteins
The click-modifications of proteins, peptides, and peptidomimetics have also been extensively studied.22 A significant proportion of this work is focused on the modification of peptide-like structures, to improve their therapeutic properties and metabolic profile.23 However, the modification of expressed proteins is also a hot application of click chemistry.The cyclization and modification of peptidic structures has been frequently examined within the context of click chemistry.24 It has also been used to enforce secondary25 and tertiary26 structures in polypeptides. With many of these protocols, the lack of good selectivity for monomer/dimer formation can be a problem due to inter-site reactions on solid supports. A method has been developed for more selective monomer cyclization by Ahsanullah and Rademann,27 but this does not make use of alkynes and is only selective for 1,5-substituted triazoles.
The ligation and cyclization of other biologically relevant molecules has also been accomplished. Oligosaccharides have been cyclized to produce cyclodextrin-like structures by Bodine,28 and, later, Chen and coworkers.29 Cyclic estrone tetramers were also produced using a combination of Glaser coupling and CuAAC by Sierra.30 The estrone tetramers were capable of including unusual functionality, including ferrocene units to form an example of a bioorganometallic conjugate.
Eichler explored the use of selective deprotection, allowing the successive ligation of individual peptidic units to a cyclic scaffold to produce trivalent scaffolded peptides.31 Aucagne and Leigh also took advantage of protection chemistry to perform one-pot ligations of multiple oligopeptides.32 Of particular interest was the mild, and highly orthogonal silver-catalyzed cleavage of trimethylsilyl groups they used,33 which allowed both ligation steps and the deprotection step to occur without purification, in the same pot. The yields were generally excellent, demonstrating that the presence of the deprotecting silvercatalyst was not noticeably detrimental to the CuAAC process.
The addition of cyclic RGDpeptides to dendrimeric structures was also accomplished, to create multivalent αVβ3 integrin antagonists.34 These dendrimers were then evaluated in vivo and found to have selective accumulation in cancerous cells when conjugated to a cyclic chelating prosthetic group, 1,4,7,10-tetraazadodecane-N,N′,N′′,N′′′-tetraacetic acid (DOTA).35 Using 111Inlabelling experiments, they determined that the number of cyclic RGD units present on the dendrimer was positively correlated with accumulation of the multimer in xenograft tumour cells in mice. The facile construction of dendrimers such as these could allow easier screening and development of multivalent therapeutic compounds based on peptidic structures.
The ability of triazoles to act as isosteres and conformationally rigid mimics of peptide bonds36 has also been exploited. Recently, Ghadiri and coworkers performed a set of binding and computational experiments to probe the binding pocket of somatostatin receptors using 16 different cyclic peptide diastereomers containing the somatostatin pharmacophore.37 The CuAAC proved vital to constructing these compounds, due to its ability to selectively produce 1,4-disubstituted triazoles, which can act as an analogue of the “trans-” amide bond, allowing the cyclic pseudotetrapeptides to act as conformationally rigid β-turn motifs. Similarly, the same group performed binding and computational studies on the interaction of HDAC with triazole-containing ampicidin derivatives (Fig. 2).38 Using both the CuAAC and a microwave-assisted thermal AAC, they were able to mimic “locked” cis- and trans-amide geometries within the cyclic pseudotetrapeptide, and determine the active conformation of the antagonist in solution. These two studies demonstrate the great utility of the triazole unit as both a pharmacophore and as a “locked” amide surrogate. Though the microwave-assisted thermal cycloaddition in the second study suffered from only 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity for the 1,5-substituted triazole, new methods for the regioselective, metal-free synthesis of these cis-peptide mimics have been developed by Rademann.39
:1 selectivity for the 1,5-substituted triazole, new methods for the regioselective, metal-free synthesis of these cis-peptide mimics have been developed by Rademann.39
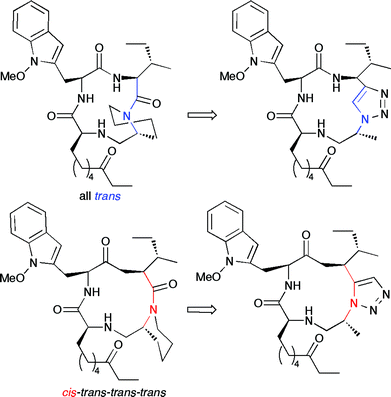 | ||
| Fig. 2 The trans-trans-trans-trans and cis-trans-trans-trans conformers of apicidin, seen on the left, can be mimicked by the triazoles seen on the right. The triazoles “lock” the molecule in the desired conformation, allowing studies of each conformer's binding to histone deacetylase. | ||
A recent study by Finn and coworkers examined the dependence of CuAACcyclization products (dimerizationvs. head-to-tail cyclization of a single monomer) upon peptide structure.40 A series of elegant experiments allowed a great insight into the mechanism of dimerization and strongly suggested that interchain hydrogen bonding is largely responsible for promoting cyclodimerization, as well as outlining a variety of factors that influence yields and selectivity in the reaction. This study should prove particularly valuable for future research in the resin-bound synthesis of cyclic peptidesvia click chemistry.
The formation of N-terminally-linked protein homo- and hetero-multimers was recently reported by Xiao and Tolbert, using a combination of native chemical ligation and click chemistry.41 Homo-dimers and -trimers were formed from the C2 domain of protein G, an immunoglobulin bindingprotein from group G streptococci bacteria, and heterodimers were formed from the aforementioned protein and C37H6, an HIV entry inhibitorpeptide. Azide- and alkyne-functionalized thioesters were added to N-terminal cysteine residues, allowing click ligation of entire expressed proteins without the need for artificial amino acid insertion. This work presents advantages over previous methods due to the relative ease of constructing heteromultimeric protein conjugates, as well as the preservation of the biochemical properties of N-terminally linked multimers compared to C-linked multimers.
The insertion of click sites into proteinsvia genetic incorporation of artificial amino acids has been accomplished by several groups. Carell and coworkers utilized the amber (UAGstop codon) suppression method in combination with expression of a Methanosarcina mazei (Mm) MSpyrrolysyl-tRNA synthetase/tRNACUA pair to insert up to three alkyne-bearing artificial amino acids into specially-evolved yellow fluorescent protein (YFP), produced by recombinant E. coli.42 Similar methods were used by Chin for the single-click modification of modified myoglobinproteins produced by recombinant E. coli.43 Alternatively, van Hest and coworkers opted to replace44 five methionine residues with azidohomoalanine in Candida antarcticalipase B, followed by click ligation of alkynyl-dansyl and bathophenanthroline residues.45 While five azidohomoalanines were present on the recombinant protein, only the residue at the N-terminus was successfully labelled.
Copper-free and in vivo click reactions
Movement away from the original dependence on copper(I) salts has also provided some fascinating new developments in bioorthogonal click chemistry.46 In particular, the work of Bertozzi and Boons, on the use of various strained cyclic alkynes to preclude the need for copper catalysis,47 has begun the use of click chemistry in in vivo applications. In particular, the use of difluorocyclooctyne derivatives48 has proven to be extremely fruitful. Other groups have shown similar results using in situ generated benzyne substrates,49 but the applicability of this reaction to living systems is negligible.A significant achievement occurred in 2010 when Bertozzi and coworkers successfully accomplished the in vivolabelling of mouse glycoproteins in splenocytes using highly strained and electronically activated unsymmetrical cyclooctyne derivatives to ligate FLAGpeptides to cell surfaces.50 The procedure involved the repeated injection of azide-modified acetylmannosamine into mice in order to allow its incorporation into cell surfaceglycoproteins as sialic acid, followed by injection of labelled cyclooctyne derivatives, like those seen in Scheme 3. While a variety of splenocytes were successfully labelled, the method presented a major drawback in that mouse serum albumin (MSA) bound strongly to the cyclooctyne derivatives, in a manner that suggests covalent bonding, possibly with cysteine residues; therefore, the bioorthogonality of these substrates is less than ideal.
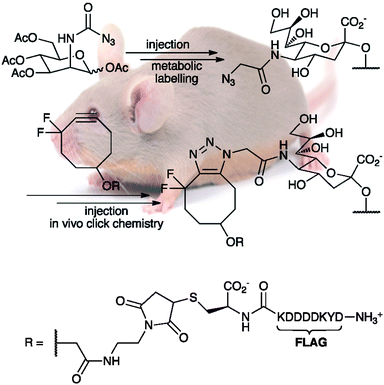 | ||
| Scheme 3 The in vivolabelling of mouse splenocyte glycoproteins with DIFO-FLAG, as performed by Bertozzi and coworkers. Use of the FLAGpeptide sequence allowed detection of the conjugates using flow cytometry, with a fluorescein isothiocyanate-labeled anti-FLAG antibody, and by Western blot procedures, using a horseradish peroxidase-ligated anti-FLAG antibody. DIFO-FLAG was found to be the most effective labelling reagent in cultured cells and the second-best for in vivo experiments. | ||
Similar work was performed in vitro by Wolfbeis and coworkers, in their use of cyclooctyne derivatives for labelling surface proteins in Chinese hamster ovarian cells.51 It is well worth noting that when the reaction was attempted in the presence of copper, significant cytotoxicity resulted.
One particularly interesting and very recent use of strained alkynes in the modification of DNA involves a slightly different cycloaddition, with a nitrile oxide in place of an azide, reported by Singh and Heaney.52 This method is distinguished from other forms of DNA click modification by its use of a ligated cyclooctyne unit; the first example of its use for DNA modificationviacopper-free click chemistry. Unlike the methods employed by Bertozzi, this reaction did not utilize unsymmetrical activated cyclooctynes, meaning that it suffered from the lack of regioselectivity inherent in the thermal AAC reaction, but for the purposes of generating a fluorescent label, this did not prove to be important. Additionally, the method involved labelling only at the 5′-phosphate moiety, implying that labelling density is limited.
An alternative strategy towards in vivo modifications has been used by other groups. Instead of eliminating copper, the groups of Finn and Wu used tris-hydroxylated and sulfonated copper(I) ligands, respectively, to enhance the water solubility of the complex and allow its use in purely aqueous solution, while at the same time preventing damage to biological molecules (Fig. 3). Finn and coworkers utilized tris(hydroxypropyltriazolyl)methylamine (THPTA) in their labelling of surface azidoglycans on cultured mammalian liver cells, and found that a 5:1 ratio of THPTA:copper(I) provided excellent protection against the generation of reactive oxygen species by the copper(I)/ascorbate combination.53 Wu et al. screened a number of ligands, and found that the inclusion of a sulfategroup and tert-butyl groups to form 2-(bis(tert-butyltriazolyl)methylamino)methyltriazolylethyl sulfate (BTTES) reached an excellent compromise between reactivity, solubility, and prevention of copper uptake by cells.54 This ligand was successfully used to effect click chemistry modification of 5-alkyne-modified fucose analogues, after their incorporation into cell-surface glycans in zebrafish embryos, allowing visualization during development with minimal cytotoxicity. Several other sub-classes of these ligands have been evaluated for activity and practicality by Finn and coworkers.55 These developments should prove especially important for research involving labelling of live cell cultures, as they preclude the need for synthesis of strained alkynes, effectively reducing the “synthetic load” of the in vitro experiments.
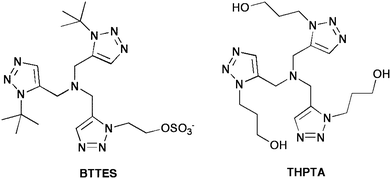 | ||
| Fig. 3 Two examples of water-soluble amine ligands used for purely aqueous click chemistry. The BTTES ligand was developed by the Wu group, and THPTA was first used by Finn. | ||
In situ click chemistry
The use of enzymes as templates for click reactions has also been investigated as an alternative to copper catalysis56 and as a method of screening compounds for pharmaceutical synthesis, offering a tantalizing opportunity to combine screening and combinatorial synthesis in one step. The first example of in situ click chemistry was reported by Sharpless in 2002, using acetylcholinesterase as a template for the construction of inhibitors. Screening 49 combinations of tacrine/phenanthridinium derivatives, a single combination produced femtomolar inhibition of the enzyme.57 The extremely tight binding was postulated to be a result not only of the bivalent nature of the synthesized compounds (phenanthridinium and tacrine are both nanomolar inhibitors of AchE), but also because of the formation of the triazole pharmacophore. The use of phenyltetrahydro-isoquinoline/tacrine combinations was also investigated by Sharpless, revealing several more femtomolar inhibitors of acetylcholinesterase.58Since the initial work by Sharpless on in situ click reactions, several other target enzymes have been investigated as templates for inhibitor synthesis. Kolb et al. examined the use of acetylenic benzenesulfonamides and a diverse set of azide derivatives as precursors to carbonic anhydrase II inhibitors,59 and Elder and Fokin similarly demonstrated the ability of HIV protease to act as a template for synthesis of a known nanomolar inhibitor.60 More recently, Omura and coworkers used an azide-modified fragment of the known chitinaseinhibitorargifin and O-propargyloximes to form novel inhibitors of chitinase in the presence of several subtypes of the enzyme.61 The synthesized 1,5-substituted triazole seen in Fig. 4 yielded a 300-fold improvement on the activity of argifin.
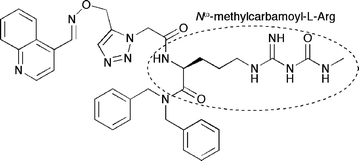 | ||
| Fig. 4 The 1,5-disubstituted triazolechitinaseinhibitor synthesized in situ by Omura and coworkers. The compound yielded double-digit nanomolar IC50 values for Serratia marcescens chitinases A and B. The outlined Nω-methylcarbamoyl-L-arginine moiety is a structural feature shared with the macrocyclic polypeptideargifin, which is responsible for forming stabilizing hydrogen bonding interactions in the active sites of many known chitinases. | ||
Recently, Finn and Miyata reported the unexpected acceleration of an in situ click reaction by histone deacetylase (HDAC).62 The reaction resulted in a high selectivity for the 1,4-substituted triazole, characteristic of copper catalysis, as well as greatly increased turnover. It was postulated that the trace presence of copper (introduced with the alkyne precursor, due to its synthesis using the Sonogashira reaction) in the enzyme active site was responsible for this selectivity and rate enhancement. Though the IC50 of the inhibitor synthesized was in the millimolar range, this discovery could potentially lead to the use of enzymes as tunablecatalytic templates for target-guided synthesis.
Click chemistry for biological imaging techniques
The area of in vivo molecular imaging is also an area that has benefited from the orthogonal reactivity of the CuAAC. Positron emission tomography (PET)63 usually relies on the labelling of biologically active molecules with 18F, with a problematic half-life of only 110 min. This problem is increasingly being overcome by the late-stage incorporation of the radiolabel into molecules,64 and click chemistry methods are beginning to see use in this field, particularly in the labelling of peptides as a method for in vivo imaging of cancer cells.65 The speed and selectivity of click reactions are highly conducive to fast and effective syntheses of 18F-labelled compounds.66 A recent example is the synthesis of 18F-labelled glycopeptides by Prante and coworkers.67 Utilizing the AAC reaction, a total synthesis time of only 75 min was achieved, with 18–20% overall radiochemical yield (RCY). The metabolic stability and distribution of these neuratensin receptor-1 (NTR) agonists were evaluated in vivo in HT-29 xenografted mice, showing merely double-digit nanomolar affinity for NTR, making the glycopeptides promising as PET imaging agents.Very recently, Andre Luxen and coworkers achieved the ligation of an 18F-labelled aryl moiety to double-stranded oligonucleotides (siRNA) using the AAC reaction.68 A total synthesis time of 120 min was realized, with an overall RCY of 15 ± 5%. This represents a considerable improvement over Inkster and coworkers' previous synthesis (the first to use CuAAC as a method for 18F-labelling of oligonucleotides with a prosthetic group) of 18F-labelled oligonucleotides which, while groundbreaking, left much to be desired with its comparatively low RCY and 276 min synthesis time.69
CuAAC has also been used for the construction of novel ligands for other radioactive nuclei. Work by Schibli and coworkers has resulted in a number of metal-chelating bioconjugates for 99mTc and the longer-lived 188Re, both of which are used for medical imaging techniques.70 The triazole unit in these compounds acts both as a ligand for the metal centre and as a linking unit between the chelating portion of the molecule and a biomolecular conjugate, such as oligonucleotides, or AZT and other azidocarbohydrates. Metzler-Nolte and coworkers also recently synthesized peptide nucleic acid-based ligands for the chelation of Tc and Retricarbonyl compounds for similar purposes.71
Finally, the copper-free ligation of quantum dots to N-azidoacetylmannosamine residues (similar to the in vivolabelling studies by Bertozzi) and their use as cell-surface glycoproteinlabelling reagents was investigated by Texier et al.72 This labelling method was found to have tremendous sensitivity, allowing visualization of cell membranes at cyclooctyne concentrations of only 250nM.
Mode B: Reaction of the alkyne C–H bond
The dimerization of terminal alkynes, known as the Glaser-coupling, the Eglinton-coupling and the Cadiot–Chodkiewicz coupling, is one of the oldest reactions in organic chemistry. Initially discovered in 1869 by Carl Glaser with the oxidative formation of diyne from copper(I) phenylacetylide and air,73 the Glaser coupling in water dated back to at least 1882, when Baeyer synthesized indigo using potassium ferricyanide as the oxidant.74 Such couplings can be performed under bio-compatible conditions. Water-soluble conjugated rotaxanes and “naked” molecular dumbbells were synthesized from such couplings in water using hydrophobic interactions to direct rotaxane formation.75Because of the slightly acidic nature of the sp C–H bonds, the reaction of metal acetylides with various electrophiles is one of the most general strategies in organic transformations.76 Traditionally, such reactions are carried out by using alkali metalacetylides, which are air- and water-sensitive. The catalytic coupling of terminal alkynes with aryl halide (the Sonogashira reaction) is a general and reliable method for connecting relatively inert terminal alkynes with inert aryl halides efficiently and it is one of the first recognized reactions for the chemical modification of biomolecules in aqueous media. In 1990, Casalnuovo and co-workers reported that unprotected iodonucleosides, iodonucleotides, and iodoamino acids underwent coupling with acetylenes catalyzed by water-soluble Pd(0) complexes Pd(PPh2(m-C6H4SO3M))3 (M = Na+, K+) in aqueous media.77 They also provided an alternative synthesis of T-505, part of a family of chain-terminating nucleotide reagents used in automated DNA sequencing and labeling, from the coupling reaction of 5-iododideoxyuridine 5′-triphosphate with the unprotected fluorescein dye in aqueous media.
Another example is the coupling of biotinyl-glutamoylpropargylamide, which is a water-soluble biotin derivative, with Pro(p-I-Phe)-bradykinin in aqueous TAPS buffer (pH 8.3) at 35 °C to give 75% yield of target product after 4 h.78 It has been shown that the specific protection for functional groups in protein was not necessary.
Ghadiri et al. reported a tri-coupling of highly charged peptides (-6 to +9) of considerable length (17–33 residues) with a trialkyne by using a Pd(0)catalyst under both acidic (pH 5.0) and basic (pH 7.5) conditions in water, efficiently giving protein-sized structures (12,000 mol wt).79 These peptides, containing amines, carboxylates, guanidines, hydroxyls, and thiolesters, do not have to be protected during the reaction. However, free thiols, thioethers, and bipyridyl moieties were shown to be non-compatible, perhaps because these functional groups serve as competing ligands for palladium as well as copper.
Granja et al. used palladium/carbon associated with 4-diphenylphosphinobenzoic acid (4-DPPBA) or the triphenyl-phosphine ligand to synthesize an alkyne-modified peptide with a pyridinyl bromide in aqueous DMF.80
The ligation of organometallic frameworks has also been accomplished thanks to bioorthogonal Sonogashira reactions, particularly in the field of bioorganometallic conjugates. The synthesis of porphyrin-functionalized oligonucleotides was reported by Stulz and coworkers, with the remarkable accomplishment of incorporating up to 11 adjacent porphyrin-labelled nucleotides,81 followed by duplex formation. While the structures had very low melting temperatures, a peculiar melting profile with two stages was seen, which implied a helical secondary structure with porphyrin stacking when the DNA–porphyrin arrays were in single-stranded form (Scheme 4).
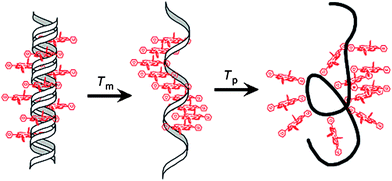 | ||
| Scheme 4 A schematic representation of the two-stage melting profile of multi-functionalized DNA–porphyrin arrays, as reported by Stulz. Image reproduced with permission from ACS Journals and the authors. | ||
Ruthenium and osmium bipyridyl functionalities were also covalently attached to nucleobases and used in primer extension to incorporate them into oligonucleotides.82 Due to the overlap of Ru2+'s oxidation potential with guanine’s, it was not deemed electrochemically useful, but these compounds displayed luminescence, allowing visualization. The Os2+ compounds had a favourable oxidation potential, and could be used for electrochemical detection. The same group also performed similar studies with ferrocene-functionalized nucleotides.83 In both cases, the Sonogashira reaction took place in aqueous/acetonitrilesolvent. These and other transformations using DNA as a supramolecular scaffold have been recently reviewed.84
Ferrocene was also conjugated to small peptide chains via a Sonogashira reaction with 4-iodophenylalanine and alkyne-functionalized ferrocene derivatives.85 The neuropeptide [Leu5]-enkephalin was thus functionalized. In combination with methods such as HPLC with electrochemical detection (HPLC-ECD), the incorporation of these organometallic frameworks into DNA and peptides allows their detection at extremely low concentrations. It is also conceivable that if utilized on recombinant proteins expressing appropriate artificial amino acids, similar work could be accomplished.
Carbon monoxide releasing compounds have also been tethered to peptides for investigation of their ability to effect localized delivery of carbon monoxide to cells. Schnatzschneider and coworkers carried out the post-functionalization of an oligopeptide sequence with the known CO-releasing molecule molybdenum tris-pyrazolylmethanetricarbonyl using both click chemistry and the Sonogashira coupling.86 The carbon monoxide releasing abilities of the molecule were largely unaffected by peptideconjugation, releasing an average of 1.7 equivalents of CO per molecule upon irradiation at 365 nm. If conjugated to peptides, such as the previously mentioned multivalent integrin antagonists, complexes such as these could prove valuable for targeted cellular delivery of CO.
Nucleophilic addition reactions
The nucleophilic addition of terminal alkynes to various unsaturated electrophiles is a classical reaction, allowing the formation of a C–C bond while simultaneously introducing the alkyne functionality. Imine and iminium formation in aqueous medium are equilibrium processes and common biological reactions in nature. The utilization of such an equilibrium in water for alkyne-nucleophilic addition would provide interesting opportunities such as peptide ligations in biological studies. However, a prerequisite of this classical reaction is the stoichiometric generation of highly reactive metal acetylides under anhydrous conditions, which are not compatible with various functionalities and biological conditions. Recently, Li and others have pioneered a wide range efficient catalytic addition of terminal alkynes in water (Scheme 5).87 These reactions are potentially amenable for reaction of biomolecules if the nucleophilic addition partner can be delivered to the desired reaction sites. One of such type of reactions is the so-called A3 (aldehyde–alkyne–amine) coupling (named by Liet al.).88Catalysts, including combined ruthenium and copper,89silver,90gold91 and ironnanoparticles,92 were developed in water under mild conditions. A potential application in biological systems was reported by Che and co-workers on the gold-catalyzed A3 reaction with proline derivatives.93 Another recent application generates butenolide derivatives by the reaction of oxylic acid, alkyne, and secondary amine.94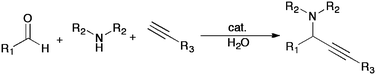 | ||
| Scheme 5 A general scheme for the aqueous A3 coupling developed by Li and coworkers. Metals utilized include silver, gold, ruthenium and copper and ironnanoparticles. Substrates have included aliphatic and aromatic aldehydes and alkynes. | ||
Conclusions
With the increasing focus of biological studies on molecular level transformations, chemical tools that are simple, easy to use, uninterrupted by the various biomolecules and functions and can proceed under mild ambient biological conditions play an increasing role. The “on-call” nature of the alkyne C–H or CC bond allows selective and triggerable reactions with a variety of substrates. Recent advances in the complex functionalization of biomolecules, as well as useful methodological developments for purely aqueous reactions, are pushing alkynes to the forefront as a versatile tool for biological applications.
Acknowledgements
We are grateful to the Canada Research Chair (Tier I) Foundation (to CJL), CFI, FQRNT and NSERC for partial support of our research.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS; V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS.
- G. A. Burley, J. Gierlich, M. R. Mofid, H. Nir, S. Tal, Y. Eichen and T. Carell, J. Am. Chem. Soc., 2006, 128, 1398–1399 CrossRef CAS.
- P. M. E. Gramlich, C. T. Wirges, J. Gierlich and T. Carell, Org. Lett., 2008, 10, 249–251 CrossRef CAS.
- J. Gierlich, K. Gutsmiedl, P. Gramlich, A. Schmidt, G. Burley and T. Carell, Chem.–Eur. J., 2007, 13, 9486–9494 CrossRef CAS.
- F. Seela and V. Sirivolu, Chem. Biodiv., 2006, 3, 509–514 CrossRef CAS.
- F. Seela, V. R. Sirivolu and P. Chittepu, Bioconjugate Chem., 2008, 19, 211–224 CrossRef CAS.
- V. R. Sirivolu, P. Chittepu and F. Seela, ChemBioChem, 2008, 9, 2305–2316 CrossRef CAS.
- P. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS.
- S. D. Brown and D. Graham, Tetrahedron Lett., 2010, 51, 5032–5034 CrossRef CAS.
- K. Gogoi, M. V. Mane, S. S. Kunte and V. A. Kumar, Nucleic Acids Res., 2007, 35, e139 CrossRef.
- G. Godeau, C. Staedel and P. Barthećlećmy, J. Med. Chem., 2008, 51, 4374–4376 CrossRef CAS.
- R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859–6864 CrossRef CAS.
- P. Kočalka, A. H. El-Sagheer and T. Brown, ChemBioChem, 2008, 9, 1280–1285 CrossRef CAS.
- S. S. Pujari, H. Xiong and F. Seela, J. Org. Chem., 2010, 75, 8693–8696 CrossRef CAS.
- M. Nakane, S. Ichikawa and A. Matsuda, J. Org. Chem., 2008, 73, 1842–1851 CrossRef CAS.
- I. K. Lee, J. D. Ahn, H. S. Kim, J. Y. Park and K. U. Lee, Curr. Drug Targets, 2003, 4, 619–623 Search PubMed.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS; J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC; Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- J. Rizo and L. M. Gierasch, Annu. Rev. Biochem., 1992, 61, 387–416 CrossRef CAS.
- S. S. van Berkel, B. van der Lee, F. L. van Delft and F. P. J. T. Rutjes, Chem. Commun., 2009, 4272–4274 RSC; J. Springer, K. R. de Cuba, S. Calvet-Vitale, J. A. J. Geenevasen, P. H. H. Hermkens, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2008, 2592–2600 CrossRef CAS; J. M. Holub, H. Jang and K. Kirshenbaum, Org. Lett., 2007, 9, 3275–3278 CrossRef CAS; J. H. van Maarseveen, W. S. Horne and M. R. Ghadiri, Org. Lett., 2005, 7, 4503–4506 CrossRef CAS; R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS; V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra and J. H. van Maarseveen, Org. Lett., 2006, 8, 919–922 CrossRef CAS.
- M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org. Chem., 2010, 446–457 CrossRef CAS.
- O. Torres, D. Yüksel, M. Bernardina, K. Kumar and D. Bong, ChemBioChem, 2008, 9, 1701–1705 CAS.
- Ahsanullah and J. Rademann, Angew. Chem., Int. Ed., 2010, 49, 5378–5382 CrossRef.
- K. D. Bodine, D. Y. Gin and M. S. Gin, Org. Lett., 2005, 7, 4479–4482 CrossRef CAS; K. D. Bodine, D. Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638–1639 CrossRef CAS.
- S. Muthana, H. Yu, H. Cao, J. Cheng and X. Chen, J. Org. Chem., 2009, 74, 2928–2936 CrossRef CAS.
- P. Ramírez-López, M. C. de la Torre, H. E. Montenegro, M. Asenjo and M. A. Sierra, Org. Lett., 2008, 10, 3555–3558 CrossRef CAS.
- R. Franke, C. Doll and J. Eichler, Tetrahedron Lett., 2005, 46, 4479–4482 CrossRef CAS.
- V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505–4507 CrossRef CAS.
- A. Orsini, A. Vitérisi, A. Bodlenner, J. Weibel and P. Pale, Tetrahedron Lett., 2005, 46, 2259–2262 CrossRef CAS.
- I. Dijkgraaf, A. Y. Rijnders, A. Soede, A. C. Dechesne, G. W. van Esse, A. J. Brouwer, F. H. M. Corstens, O. C. Boerman, D. T. S. Rijkers and R. M. J. Liskamp, Org. Biomol. Chem., 2007, 5, 935–944 RSC.
- L. M. De León-Rodríguez and Z. Kovacs, Bioconjugate Chem., 2008, 19, 391–402 CrossRef CAS.
- V. D. Bock, D. Speijer, H. Hiemstra and J. H. van Maarseveen, Org. Biomol. Chem., 2007, 5, 971–975 RSC.
- J. Beierle, W. Horne, J. van Maarseveen, B. Waser, J. Reubi and M. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4725–4729 CrossRef CAS.
- W. Horne, C. Olsen, J. Beierle, A. Montero and M. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4718–4724 CrossRef CAS.
- Ahsanullah, P. Schmieder, R. Kühne and J. Rademann, Angew. Chem., Int. Ed., 2009, 48, 5042–5045 CrossRef.
- R. Jagasia, J. M. Holub, M. Bollinger, K. Kirshenbaum and M. G. Finn, J. Org. Chem., 2009, 74, 2964–2974 CrossRef CAS.
- J. Xiao and T. J. Tolbert, Org. Lett., 2009, 11, 4144–4147 CrossRef CAS.
- E. Kaya, K. Gutsmiedl, M. Vrabel, M. Müller, P. Thumbs and T. Carell, ChemBioChem, 2009, 10, 2858–2861 CrossRef CAS.
- D. P. Nguyen, H. Lusic, H. Neumann, P. B. Kapadnis, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 8720–8721 CrossRef CAS.
- A. J. Link and D. A. Tirrell, Methods, 2005, 36, 291–298 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. V. Eldijk and J. C. M. V. Hest, Bioconjugate Chem., 2008, 19, 1127–1131 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS; J. M. Baskin and C. R. Bertozzi, Aldrichimica Acta, 2010, 43, 15–23 CAS; J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS; X. Ning, J. Guo, M. Wolfert and G. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- E. M. Sletten, H. Nakamura, J. C. Jewett and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 11799–11805 CrossRef CAS.
- F. Zhang and J. E. Moses, Org. Lett., 2009, 11, 1587–1590 CrossRef CAS; F. Shi, J. P. Waldo, Y. Chen and R. C. Larock, Org. Lett., 2008, 10, 2409–2412 CrossRef CAS.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS.
- P. Kele, X. Li, M. Link, K. Nagy, A. Herner, K. Lőrincz, S. Béni and O. S. Wolfbeis, Org. Biomol. Chem., 2009, 7, 3486–3490 RSC.
- I. Singh and F. Heaney, Chem. Commun., 2011, 47, 2706–2708 RSC.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS.
- D. Soriano del Amo, W. Wang, H. Jiang, C. Besanceney, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef.
- S. I. Presolski, V. Hong, S. Cho and M. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS.
- S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC; K. B. Sharpless and R. Manetsch, Expert Opin. Drug Discovery, 2006, 1, 525–538 Search PubMed; X. Hu and R. Manetsch, Chem. Soc. Rev., 2010, 39, 1316–1324 RSC.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CAS; R. Manetsch, A. Krasiński, Z. Radić, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2004, 126, 12809–12818 CrossRef CAS.
- A. Krasiński, Z. Radić, R. Manetsch, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2005, 127, 6686–6692 CrossRef CAS.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Röper, K. B. Sharpless, C. Wong and H. C. Kolb, Angew. Chem., Int. Ed., 2005, 44, 116–120 CrossRef CAS.
- M. Whiting, J. Muldoon, Y. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 1435–1439 CrossRef CAS.
- T. Hirose, T. Sunazuka, A. Sugawara, A. Endo, K. Iguchi, T. Yamamoto, H. Ui, K. Shiomi, T. Watanabe, K. B. Sharpless and S. Omura, J. Antibiot., 2009, 62, 277–282 CrossRef CAS.
- T. Suzuki, Y. Ota, Y. Kasuya, M. Mutsuga, Y. Kawamura, H. Tsumoto, H. Nakagawa, M. Finn and N. Miyata, Angew. Chem., Int. Ed., 2010, 49, 6817–6820 CrossRef CAS.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS.
- T. Furuya, J. Klein and T. Ritter, Synthesis, 2010, 1804–1821 CAS; T. Ritter, Nature, 2010, 466, 447–448 CrossRef CAS; P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150–12154 CrossRef CAS.
- Z. Li, Z. Wu, K. Chen, F. T. Chin and X. Chen, Bioconjugate Chem., 2007, 18, 1987–1994 CrossRef CAS; J. Marik and J. L. Sutcliffe, Tetrahedron Lett., 2006, 47, 6681–6684 CrossRef CAS.
- T. L. Ross, Curr. Radiopharm., 2010, 3, 202–223 Search PubMed.
- S. Maschauer, J. Einsiedel, R. Haubner, C. Hocke, M. Ocker, H. Hubner, T. Kuwert, P. Gmeiner and O. Prante, Angew. Chem., Int. Ed., 2010, 49, 976–979 CAS.
- F. Mercier, J. Paris, G. Kaisin, D. Thonon, J. Flagothier, N. Teller, C. Lemaire and A. Luxen, Bioconjugate Chem., 2011, 22, 108–114 CrossRef CAS.
- J. A. H. Inkster, M. J. Adam, T. Storr and T. J. Ruth, Nucleosides, Nucleotides Nucleic Acids, 2009, 28, 1131–1143 CrossRef CAS.
- T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwé and R. Schibli, J. Am. Chem. Soc., 2006, 128, 15096–15097 CrossRef CAS.
- G. Gasser, A. M. Sosniak, A. Leonidova, H. Braband and N. Metzler-Nolte, Aust. J. Chem., 2011, 64, 265–272 CrossRef CAS.
- A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffeć and I. Texier, Bioconjugate Chem., 2010, 21, 583–588 CrossRef CAS.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1882, 15, 50–56 CrossRef.
- For reviews, see: P. Cadiot, and W. Chodkiewicz, in Chemistry of Acetylenes, ed. H. G. Viehe, Marcel Dekker, New York, 1969, pp. 597 Search PubMed.
- L. Brandsma in Preparative Acetylenic Chemistry, ed. Heus-Y. A. Kloos, R. Van Der Heiden, H. D. Verkruijsse, Elsevier, New York, 2nd edition, 1988 Search PubMed; L. Brandsma, S. F. Vasilevsky and H. D. Verkruijsse inApplication of Transition Metal Catalysts in Organic Synthesis, Springer-Verlag, New York, 1999 Search PubMed.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324–4330 CrossRef.
- A. Hessler, O. Stelzer, H. Dibowski, K. Worm and F. P. Schmidtchen, J. Org. Chem., 1997, 62, 2362–2369 CrossRef.
- D. T. Bong and M. R. Ghadiri, Org. Lett., 2001, 3, 2509–2511 CrossRef CAS.
- R. J. Brea, M. P. Lopez-Deber, L. Castedo and J. R. Granja, J. Org. Chem., 2006, 71, 7870–7873 CrossRef CAS.
- L. Fendt, I. Bouamaied, S. Thöni, N. Amiot and E. Stulz, J. Am. Chem. Soc., 2007, 129, 15319–15329 CrossRef CAS.
- M. Vrábel, P. Horáková, H. Pivoňková, L. Kalachova, H. Černocká, H. Cahová, R. Pohl, P. Šebest, L. Havran, M. Hocek and M. Fojta, Chem.–Eur. J., 2009, 15, 1144–1154 CAS.
- P. Brázdilová, M. Vrábel, R. Pohl, H. Pivoňková, L. Havran, M. Hocek and M. Fojta, Chem.–Eur. J., 2007, 13, 9527–9533 CrossRef CAS.
- T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138–148 RSC.
- U. Hoffmanns and N. Metzler-Nolte, Bioconjugate Chem., 2006, 17, 204–213 CrossRef CAS.
- H. Pfeiffer, A. Rojas, J. Niesel and U. Schatzschneider, Dalton Trans., 2009, 4292–4298 RSC.
- C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS.
- C. Wei, Z. Li and C.-J. Li, Synlett, 2004, 1472–1483 CAS; L. Zani and C. Bolm, Chem. Commun., 2006, 4263–4275 RSC.
- C.-J. Li and C. Wei, Chem. Commun., 2002, 268–269 RSC.
- C. Wei, Z. Li and C.-J. Li, Org. Lett., 2003, 5, 4473–4475 CrossRef CAS.
- C. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS.
- T. Zeng, W. Chen, C. M. Cirtiu, A. Moores, G. Song and C.-J. Li, Green Chem., 2010, 12, 570–573 RSC.
- V. K. Y. Lo, K. K. Y. Kung, M. K. Wong and C. M. Che, J. Organomet. Chem., 2009, 694, 583–591 CrossRef CAS.
- Q. Zhang, M. Cheng, X. Hu, B. G. Li and J. X. Ji, J. Am. Chem. Soc., 2010, 132, 7256–7257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |