Carbocatalysis: Heterogeneous carbons finding utility in synthetic chemistry
Daniel R.
Dreyer
and
Christopher W.
Bielawski
*
Department of Chemistry and Biochemistry, The University of Texas at Austin, 1 University Station, A5300, Austin, TX 78712, USA. E-mail: bielawski@cm.utexas.edu; Tel: +1-512-232-3839
First published on 14th April 2011
Abstract
In this minireview, we discuss the utility of heterogeneous carbons as catalysts for facilitating a broad range of synthetic transformations. While such materials are commonly used as supports for transition metals that are catalytically active, carbons that are free of metals are also capable of enabling useful chemical reactions. Carbon catalysts hold promise in the development of sustainable alternatives to existing metal-dependent processes, as well as the discovery of mechanisms and transformations that are altogether new. Spanning from the 1930s to the present day, we provide a broad overview of the utility of carbon to facilitate various oxidation, reduction, and bond forming processes. Lastly, we will present some challenges for the future of the field.
 Daniel R. Dreyer | Daniel R. Dreyer is a Ph.D. candidate at The University of Texas at Austin in the research group of Prof. C. W. Bielawski. He received his B.S. in Chemistry in 2007 from Wheaton College (IL), where he conducted research in confocal microscopy with Prof. Daniel L. Burden. He also studied X-ray reflectometry and plasma polymerization with Prof. Mark D. Foster at the University of Akron. His research interests include applications of ionic liquids in synthetic polymer chemistry, structurally dynamic materials, carbocatalysis, and novel electrolytes for graphene-based energy-storage devices. |
 Christopher W. Bielawski | Christopher W. Bielawski received a B.S. in Chemistry from the University of Illinois at Urbana-Champaign (1996) and a Ph.D. from the California Institute of Technology (2003). After postdoctoral studies (also at Caltech), he launched his independent career at The University of Texas at Austin in 2004, where he is currently a Professor of Chemistry. His research program is synthetic in nature and lies at the interface of organic, polymer, and materials chemistry. |
Introduction
While recent interest in carbon has focused primarily on graphene,1carbon nanotubes (CNTs)2 and fullerenes,3 other materials of similar composition have a long history, a significant portion of which has been devoted to understanding their role in synthetic chemistry. Carbon is widely available and inexpensive,4 two features that have facilitated the broad exploration and utility of this material in synthesis. Moreover, the chemical and physical properties of any given carbon depend strongly on its source and method of preparation.5–7 Naturally-occurring carbons may be categorized into two classes: (1) those that are well-defined, such as lamellar graphite,8 and (2) those that are ill-defined, such as glassy or amorphous carbons,9–11 which lack long range order and uniform hybridization. To complement these natural materials, which are abundant, a wide range of synthetic variants has also been developed, including charcoal (the char recovered after pyrolyzing an organic precursor)9,12 and activated carbons (porous carbons treated with chemical reagents to increase their adsorptive properties),6,9 and many are now commercially prepared on large scales.5 Most, if not all of these materials have found utility in the growing field of “carbocatalysis”, as will be described below. While carbons have also found extensive use as supports for catalytically active transition metals, such applications are beyond the scope of the current discussion and we direct readers to other recent reviews on these topics.6,7,13,14Metal-free catalytic applications of carbon
The use of metal-free, heterogeneous carbons as catalysts in synthetic reactions (which we will refer to herein as carbocatalysis15) may be traced back to at least the 1930s, when Kutzelnigg and Kolthoff independently demonstrated that activated charcoals are capable of catalyzing the aerobic oxidation of ferrocyanide to ferricyanide (Scheme 1).16,17 The authors of these studies reasoned that the acidic surface generated via thermal activation of the carbon improved adsorption of gaseous oxygen (compared to unactivated carbons), promoting the oxidation reaction. For example, when the charcoal was heated to 900 °C, the resulting material was hydrophobic;17–21 however, when a lower activation temperature was employed (e.g., 400 °C), the carbon was more hydrophilic and interacted strongly with basic solutions, consistent with the presence of acidic functional groups decorated on the carbon surface.†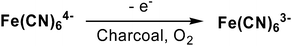 | ||
| Scheme 1 Activated charcoal catalyzed aerobic oxidation of ferrocyanide to ferricyanide. | ||
During this same time period, Rideal and Wright also investigated the ability of charcoals to catalyze the aerobic oxidation of oxalic acid (1), among other substrates. Consistent with the charcoal being the active catalyst, no conversion was observed in the absence of a carbon at the relatively low temperatures employed in the reactions studied (30–100 °C).22–24 It was suggested by the authors of this study that the carbon used in such reactions underwent aerobic oxidization and afforded a material decorated with surface-bound geminal diols. Upon further reaction with ambient oxygen, the diols formed peroxide intermediates, which then reacted with 1 to form carbon dioxide and water (Scheme 2). Malonic acid was found to undergo similar oxidation processes, although glyoxylic acid was obtained as a product. In contrast, monofunctional acids containing a single polar group (e.g., formic acid, acetic acid, fatty acids, etc.) were found to be unreactive under these conditions.22 Although the authors offered no direct explanation for this lack of reactivity, we surmise that monofunctionalized species may adsorb too weakly to the surface, disabling further reaction.
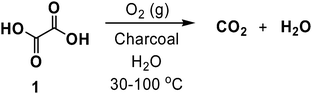 | ||
| Scheme 2 Charcoal catalyzed, aerobic oxidation of oxalic acid. | ||
Reaction scope of carbocatalysts
High surface area carbons are particularly useful in carbocatalytic applications because of their high ratio of catalytically active surface area to weight.7 For example, commercially available carbon molecular sieves (CMSs), which are primarily used for the separation of small molecules in the gas phase,25,26 have been prepared with exceptionally high surface areas, reaching over 3000 m2 g−1 in some instances.27 When CMSs were incorporated into flow reactors operating at 230 °C (using air as the carrier gas at 5 mL min−1), gaseous 1-propanol, 2-propanol, and propanal (introduced at 0.2 mL h−1) were found to oxidize to their respective aldehyde and ketone products (Table 1). However, when the carrier gas was switched to nitrogen, alcohol oxidation ceased within 2 h; reactivity rapidly resumed upon the re-introduction of air. A dependence on an oxygen-rich atmosphere is consistent with the alcohol-mediated reduction of the active sites on the surfaces of the CMSs, which were then regenerated by oxygen to complete a catalytic cycle.| Substrate | Products | Selectivity (%) | Conversion (%) |
|---|---|---|---|
| 1-Propanol | Propene | 63 | 40 |
| Propanol | 13 | ||
| 2-Propanol | Propene | 70 | 46 |
| Acetone | 20 | ||
| Propanal | Acetaldehyde | 69 | 48 |
| Ethanol | 10 | ||
| Ethylene | 8 |
There are at least two mechanisms that may rationalize the products of the aforementioned processes. The first involves the reaction between an alcohol and carbocations proposed to be present on the CMS surface.27 Similar to inorganic oxides,28,29 a Lewis acid catalyzed process should afford the aldehyde product viahydride abstraction followed by deprotonation of the alcohol. Consistent with this mechanism, acetaldehyde was obtained from ethanol in the aforementioned CMS catalyzed process. Since the CMS may also function as a Brønsted acid, a second possible mechanism may involve alcohol dehydration. For example, as shown in Table 1, various olefinic products were obtained from their respective C3alcohols.
In another example of a carbon catalyzed oxidation process, Ritter and co-workers determined that 4-chlorophenol may undergo oxidative cleavage in the presence of graphite and hydrogen peroxide to afford carbon dioxide, water, and hydrochloric acid.30 The reaction was believed to proceed via the graphite-catalyzed decomposition and activation of hydrogen peroxide, similar to the reactivity observed when Fenton's reagent is used as an oxidant.31
While most of the reactions involving carbocatalysts are oxidations (typically involving atmospheric oxygen32 or hydrogen peroxide30,33 as the terminal oxidants), carbon catalyzed reductions have also been demonstrated. For example, Cho and co-workers reported in 1985 that graphite catalyzes the reduction of nitroarenes (2) to their corresponding anilines (3) in 85–98% conversion using hydrazine hydrate as the terminal reductant (Scheme 3).34 The reduction of nitrobenzene to aniline is a four-electron process, while hydrazine commonly functions as a two-electron reductant, indicating that phenylhydroxylamine may be generated as an intermediate.35 The graphite was believed to facilitate substrate adsorption, and subsequent electron transfer between the hydrazine and the nitroarene.35 Supporting this conclusion, no conversion to aniline was observed in the absence of either the graphite or the hydrazine.35,36 Moreover, the carbon catalyzed reduction of nitro groups may be a broadly applicable process as aliphatic species (e.g., nitroethane) were also converted to their respective amines (e.g., ethylamine) in similar isolated yields as aniline obtained from nitrobenzene.34
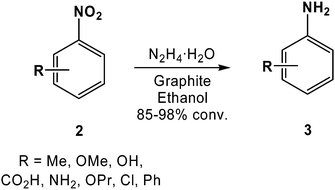 | ||
| Scheme 3 Graphite catalyzed reduction of substituted nitrobenzenes to their corresponding anilines using hydrazine hydrate as the terminal reductant. | ||
Analogous reductions have also been performed utilizing C60 and C70 (Scheme 4).37 Gaseous hydrogen was used as the terminal reductant, rather than hydrazine, and the reaction was promoted by irradiation with UV or visible light, allowing for hydrogen pressures as low as 1 atm to be used. Cooperative effects between neutral and anionic fullerenes (prepared by reacting the neutral fullerene with a nickel–aluminium alloy) were observed which resulted in improved yields of the aniline product. In order to determine if metals were the catalytically active species, the authors used inductively coupled plasma mass spectrometry (ICP-MS). Trace amounts (0.1–67.4 μg g−1) of cobalt, chromium, copper, iron, and silver were detected, with iron being the most prevalent species; palladium, platinum, and nickel were below the detection limit. However, speculation exists as to whether the reactivity observed in this study was indeed due to the fullerene.38 For example, van Bokhoven and co-workers reported that reduced C60 (C60−1; prepared as described above) was contaminated with 400 μg of nickel per gram of carbon, and C60−1 prepared using sodium naphthalenide was catalytically inactive, suggesting that nickel was the active catalyst in the aforementioned reduction reaction.
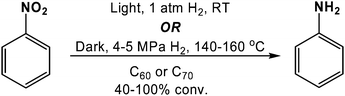 | ||
| Scheme 4 C60 or C70 catalyzed reduction of nitrobenzene to aniline using hydrogen as the terminal reductant. | ||
Fatty acids (e.g., palmitic and oleic acids) have also been reduced by heating the reactants in the presence of activated carbon and supercritical water (or near-supercritical water) at temperatures ranging from 330 °C to 385 °C (Tc = 374 °C).39 These conditions were found to facilitate decarboxylation of the acid moiety, as well as reduction of internal olefins, if present, affording linear, fully saturated alkanes one carbon shorter than their starting material. For example, palmitic acid (a saturated fatty acid) was cleaved to pentadecane in 33% conversion, while oleic acid (an omega-9 unsaturated fatty acid) was reduced and cleaved to heptadecane in 80% conversion (Scheme 5), as determined by gas chromatography. Further degradation to C8–C14alkanes was observed in small amounts (<1 mol%) at elevated temperatures (>370 °C). While the active reductant, as well as the overall mechanism, remains unclear, it was reasoned that other fatty acids, or the solvent (water), served as the terminal reductant.39
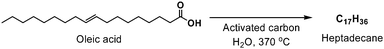 | ||
| Scheme 5 Reduction and decarboxylation of oleic acid to heptadecane using activated carbon and near- or supercritical water. | ||
In order to characterize the activation parameters of the reaction, the authors used batch reactors to perform isothermal calorimetric analyses. The experiments revealed pseudo-first order reaction kinetics (with respect to the activated carbon loading), and an activation energy of 125 ± 3 kJ mol−1 for the hydrothermal decarboxylation of palmitic acid. For comparison, an activation energy of 79 kJ mol−1 was measured for the decarboxylation of palmitic acid using Pt/C,40 a value that reflects the fact that Pt/C is a more energy efficient decarboxylation catalyst than the carbon support alone.39
Beyond functional group transformations, carbon has also found utility in the formation of more complex structures. For example, natural flake graphite has been used to catalyze the cleavage of alkyl ethers (4) using acyl halides (5) in 67–97% yield, as determined by gas-liquid chromatography (Scheme 6).41 In this study, the authors observed that tertiary alkyl ethers were selectively cleaved in the presence of secondary ethers, and that primary and secondary alkyl ethers were inert under the reaction conditions explored. Based on these results, the authors suggested a mechanism similar to that proposed for the iron(III) chloride-acetic anhydride system, where O-acylation of the ether is followed by C–O cleavage to afford a stable carbonium ion. Alternatively, nucleophilic displacement by acetate has been proposed to occur upon oxonium ion formation.42 The authors reasoned that the graphite served to stabilize the acylinium cation as well as the cation intermediate via cation-π interactions. Similar reactivity was observed in the graphite catalyzed Friedel–Crafts-type substitutions between various acid halides or alkyl halides (6) and electron rich arenes (7) to afford alkyl arenes (8) in 38–100% yield (Scheme 7). As a testament to the ability of carbons to enhance reactivity, the aforementioned acylations were found to proceed without the addition of strong Lewis acids.43,44
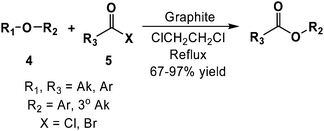 | ||
| Scheme 6 Graphite catalyzed cleavage of ethers, followed by coupling with an acyl halide (adapted from ref. 41). | ||
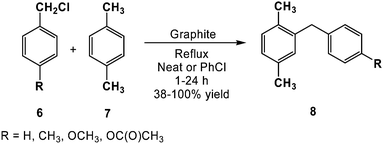 | ||
| Scheme 7 Graphite catalyzed Friedel–Crafts-type substitutions (adapted from ref. 44). | ||
Beyond its high surface area and unique chemical properties, the high thermal conductivity of graphite (19.1 W cm−1 K−1 at 300 K45) has enabled its use as a solid state thermal conductor to promote synthetic reactions. For example, the [4 + 2] cycloaddition between anthracene (9) and various electron deficient dienophiles, including diethyl fumarate (10), maleic anhydride (11) and dimethyl acetylenedicarboxylate (12), was facilitated upon physisorption of these substrates onto graphite, followed by exposure to microwave irradiation (Scheme 8).46,47 Similar reactivity was also observed in the carbonyl-ene cyclization of (+)-citronellal (13) to (+)-(14) and (−)-isopulegol (15) (isolated in a ratio of 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 from enantiopure (+)-citronellal) (Scheme 9).47 Although comparable yields of products were obtained when the aforementioned cyclization reactions were performed under conventional heating (typically under reflux in xylenes, dioxane, or chloroform), the reaction times were considerably longer (hours, as compared to minutes under microwave irradiation).
:30 from enantiopure (+)-citronellal) (Scheme 9).47 Although comparable yields of products were obtained when the aforementioned cyclization reactions were performed under conventional heating (typically under reflux in xylenes, dioxane, or chloroform), the reaction times were considerably longer (hours, as compared to minutes under microwave irradiation).
![Graphite catalyzed [4 + 2] cycloadditions between anthracene and various electron deficient dienophiles.](/image/article/2011/SC/c1sc00035g/c1sc00035g-s8.gif) | ||
| Scheme 8 Graphite catalyzed [4 + 2] cycloadditions between anthracene and various electron deficient dienophiles. | ||
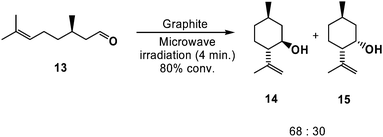 | ||
| Scheme 9 Graphite-catalyzed ene cyclization of (+)-citronellal to (±)-isopulegol. | ||
Surface functionalized carbocatalysts
The use of heterogeneous, metal-free carbons in lieu of metal catalysts in synthetic chemistry has continued to progress over the last decade, in part as a result of diminishing supplies of rare earth metals used in common industrial processes, such as the oxidative dehydrogenation of ethylbenzene to form styrene.48–51 Additionally, the discovery and development of novel forms of carbon (e.g., CNTs, graphene, fullerenes, etc.) has led to their exploration for use as catalysts.39 For example, in 2008, Su and coworkers demonstrated that oxidized CNTs are capable of catalyzing the oxidative dehydrogenation of n-butane (16) to 1-butene (17), a reaction typically performed at high temperatures (500–900 °C) over complex metal oxides (Scheme 10).52–54 As hypothesized in some of the previously described examples, atmospheric oxygen was believed to serve as the terminal oxidant in these reactions. Regardless, the interaction of the alkane with the CNT surfaces (which may have been enhanced by the presence of the oxygen functional groups), resulting in the evolution of carbon monoxide, carbon dioxide, and water, in addition to the desired olefinic product in 6.7% yield. The product yield was successfully increased (to 13.8%) through the addition of phosphorus, which may have passivated surface defects present on the oxidized CNTs.54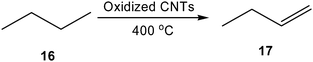
Graphite oxide facilitates a broad range of transformations
Particularly useful functionalized carbons include graphite oxide (GO) and graphene oxide, which are frequently used as precursors to obtain graphene-like materials.55,56 Over its long history,55 GO has been prepared using a variety of techniques, though the most common are embodied in the Hummers and Brodie/Staudenmaier methods. The former, first described in 1958,57 involves treating graphite (natural or synthetic) with potassium permanganate and sodium nitrate in concentrated sulfuric acid. The reaction is subsequently quenched through the addition of aqueous hydrogen peroxide, and the product is isolated either viafiltration or centrifugation, followed by washing with water or organic solvents to remove residual reagents. Like the Hummers method, the Brodie58 and Staudenmaier59 methods for preparing GO from graphite utilize strong acids (e.g., sulfuric or nitric acid), though potassium or sodium chlorate are used in lieu of potassium permanganate. Regardless of the method used, the graphite transitions from being a hydrophobic to a hydrophilic material as the oxidation proceeds. As a result of its enhanced hydrophilicity, it has been shown that water readily intercalates into the surface-functionalized GO, significantly altering its stacking structure. For example, interlamellar d-spacings were measured to increase from approximately 3.5 Å to as much as 8 Å, depending on the relative humidity.60,61While the structure of GO is still under debate,55 the Hummers, Brodie, and Staudenmaier methods of preparing GO all introduce a wide range of oxygen functional groups to the basal plane of the graphene layers as well as their periphery. The most widely adopted structural model of GO is that proposed by Lerf, Klinowski and co-workers, wherein the surface functionality is dominated by epoxides and alcohols, and the periphery is decorated with carboxylic acids (Scheme 11).62–65 Variations of this structure do exist, however, such as that proposed by Dékány and co-workers, where the oxygen functional groups are present primarily as bridging 1,3-ethers, alcohols, and quinoidal moieties.66
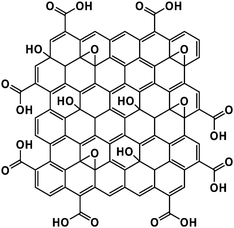 | ||
| Scheme 11 Proposed structure of graphene oxide according to the Lerf–Klinowski model.62–64 | ||
As a highly oxidized and thermodynamically unstable material,67 GO has been commonly used as a precursor to graphene and other similarly exfoliated carbons.68 Though other methods have been developed to directly exfoliate graphite into graphene,69 the use of GO as an intermediate takes advantage of the decreased interlamellar, non-covalent bonding, owing to GO's extensive surface functionalization. That is, the increased d-spacing between the layers significantly reduces the strength of the intermolecular forces responsible for graphite's stacked structure. After exfoliation of the layers, which is often accomplished using sonication or mechanical stirring, a reductant is added to the resulting dispersion. Hydrazine hydrate remains one of the most common,70 but a wide range of reductants have been used, including anhydrous hydrazine,71 borohydride,72,73hydroquinone,74,75 and other reagents.76–79
Seeking to harness GO's high chemical potential as a means to facilitate useful chemical transformations, we recently showed that GO and graphene oxide are capable of oxidizing a broad range of alcohols to their aldehyde and ketone products. For example, as summarized in Scheme 12, benzyl alcohol (18) was oxidized to benzaldehyde (19) in near quantitative conversion (>98%, as determined by 1H NMR spectroscopy). Likewise, as shown in Table 2, a variety of other primary and secondary alcohols were oxidized to their corresponding products.15,80,81 These oxidization reactions were found to be inhibited when performed under an atmosphere of nitrogen, which suggested to us that atmospheric oxygen may be the terminal oxidant. Moreover, no reactivity was observed when hydrazine-reduced graphene oxide70 or natural flake graphite were substituted for GO, indicating that the oxygen containing functional groups bound to the surface were necessary for the observed reactivity.
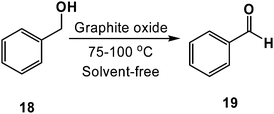 | ||
| Scheme 12 Aerobic oxidation of benzyl alcohol as facilitated by graphite oxide (GO). | ||
| Entry | Alcohol | Product | Conversion |
|---|---|---|---|
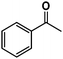
| 1 |
|
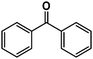
|
>98% |
| 2 |
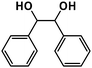
|
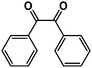
|
96% |
| 3 |
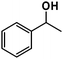
|
|
26% |
| 4 |
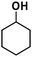
|
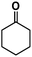
|
>98% |
| 5 |
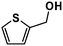
|
|
18% |
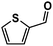
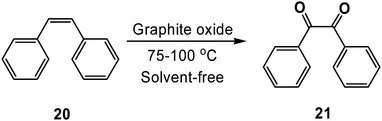
While exploring the scope of reactivity exhibited by GO, we discovered that the material was capable of facilitating Wacker-like oxidations of cis-stilbenes (20) to their corresponding benzil products (21),82 a process often requiring selenium,83cerium,84 or chromium85 species (Scheme 13). Remarkably, trans-stilbene and other trans-substituted olefins, as well as other cis-substituted olefins (e.g., cyclohexene, cis-β-methylstyrene) did not undergo significant oxidation under these conditions.15 We surmised that the stereochemistry and substitution pattern (i.e., diaryl functionalization) of cis-stilbene facilitated its reaction with GO in a manner that involved an unencumbered, favorable interaction with the carbon surface.
 | ||
| Scheme 13 Oxidation of cis-stilbene as facilitated by graphite oxide (GO). | ||
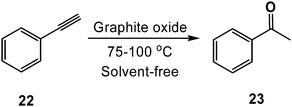
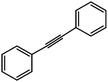
To our surprise, we also discovered that GO was capable of hydrating both alkyl and aryl alkynes (22) (Scheme 14) to their corresponding methyl ketones (23), a process that typically requires oxymercuration chemistry.15,86 A range of terminal and internal alkynes were successfully hydrated, though aryl substitution significantly enhanced the isolated yields (e.g., quantitative conversion of phenylacetylene to acetophenone was observed) (see Table 3). The observed reactivity is likely a result of GO's high acidity in aqueous media (pH = 4.5 at 0.1 mg mL−1).66 While the loadings of GO used in some of the aformentioned reactions were high (typically 100–200 wt%, relative to the substrate), the temperatures required to achieve appreciable conversions were reasonably low (≤100 °C), and GO is relatively inexpensive to prepare (<$1 g−1) and may be reused.15 As expected, the carbon material recovered was found to have undergone significant deoxygenation, as determined by FT–IR spectroscopy, powder conductivity measurements, and elemental combustion analysis.15,81 The reduction may be due, in part, to consumption of the oxygen functional groups during the reaction with the substrate, though it also likely reflects a competing thermal degradation process87 that mandates a high loading of GO.
 | ||
| Scheme 14 Hydration of phenylacetylene as facilitated by graphite oxide (GO). | ||
| Entry | Alkyne | Product | Conversion |
|---|---|---|---|
| 1 |
|
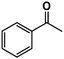
|
>98% |
| 2 |
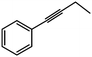
|
|
52% |
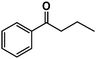
| 3 |
|
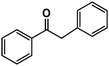
|
41% |
| 4 |
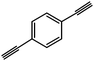
|
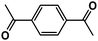
|
26% |
| 5 |
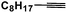
|
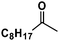
|
27% |
The aforementioned GO-catalyzed alcohol oxidations and alkyne hydrations have also been combined in an auto-tandem fashion88 to form more complex substrates, such as chalcones (24) (Scheme 15), in a single reaction vessel.89 Demonstrating the versatility of GO to facilitate multiple mechanistically-distinct reactions, both electron rich (e.g., p-methoxy substituted) and electron poor (e.g., p-nitro substituted) aryl alkynes or methyl ketones were successfully coupled with similarly electron rich and electron poor aryl alcohols or aldehydes to form the targeted products, typically in >60% isolated yields. The condensation of the methyl ketones with the aldehydes was believed to proceed via a Claisen-Schmidt-type process, where the GO acted as an acid catalyst.90 When an alkyne was substituted for the methyl ketone, however, the condensation was likely preceded by hydration of the alkyne. Likewise, when an alcohol was substituted for the aldehyde, the condensation was preceded by oxidation of the alcohol. All of these reactions were successfully combined into a single reaction vessel, which enabled the first example of forming chalcones from the coupling of alkynes and alcohols. As in many of the examples described above, the heterogeneous nature of the reaction mixtures employed facilitated isolation of the desired product(s). For example, the chalcones synthesized using GO were easily dissolved into organic solvents, such as CH2Cl2, at the conclusion of the reaction, and separated.
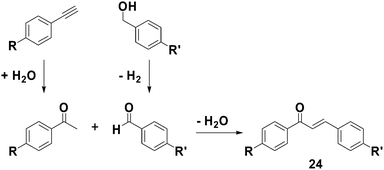 | ||
| Scheme 15 Summary of various graphite oxide facilitated coupling reactions used in the formation of chalcones (R = H, OMe, NO2). | ||
Metal-free catalysts?
A recent trend in synthetic chemistry has focused on the exploration and development of metal-free variations of transformations that typically require transition metals (e.g., aryl coupling reactions).91–95 In some cases, however, the reactions that were presumed to be metal-free were subsequently found to be catalyzed by transition metal contaminants present at trace levels, often parts-per-billion (ppb), or less.96,97 Given that most carbons are derived from naturally-occurring carbon deposits (principally highly ordered graphite, or unstructured, glassy, or amorphous carbons),5 there is potential for contamination by naturally occurring metals in many of the carbons described above. Moreover, functionalized derivatives, such as GO, often require metal-containing reagents (e.g., potassium permanganate)57 in their preparation, which may lead to further contamination. In an effort to exclude the possibility of metal-mediated catalysis, the GO described in the aforementioned alcohol and olefin oxidation and alkyne hydration studies was extensively examined using elemental combustion analysis, atomic absorption (AA) spectroscopy, and inductively coupled plasma mass spectrometry (ICP-MS).15,81,89 In all cases, manganese and other metal contaminants, such as iron, lead, aluminum and barium, were found to be present at background levels (typically on the order of a few tens of ppb). Hence, while the possibility of metal-mediated contributors cannot be excluded, functional group-free carbons (e.g., hydrazine-reduced GO and graphite) were found to be inert under identical reaction conditions. Regardless, in view of the potential pitfalls associated with contaminants, it is advisable for those working in the field of carbocatalysis to perform appropriate analyses and control experiments, in order to conclusively determine the source of the observed reactivity.Conclusions and outlook
Carbons have found broad use in many areas, such as in the reinforcement of polymer composites,98 pigments,99water purification,100 and gas storage.101 However, more recently these materials have been shown to be useful in synthesis and catalysis. While carbons have long been used as supports for metal catalysts,6,7 they have also found extensive utility as metal-free carbocatalysts for facilitating a wide range of synthetically-useful transformations. Many of the early studies focused on simple redox processes, but the field has progressed to demonstrate that carbons can facilitate more sophisticated reactions, including complex functional group transformations and carbon-carbon or carbon-heteroatom bond formations. With this increased reaction scope in view, as well as the practical advantages associated with heterogeneous catalysts,102–104 carbons may find broad applicability in synthetic chemistry, particularly in reactions that rely on increasingly scarce, metal-based catalysts. Given its long history and the extensive scope of demonstrated reactivity, the field appears to be poised for widespread adoption, as well as further exploration.One area worth commending for future study is the use of functionalized carbons, as opposed to highly pure carbons. While functional group-rich materials such as GO exhibit broad reactivity under mild conditions, their structure is not fully understood,55 which, combined with the heterogeneous nature of the catalysts employed, renders mechanistic elucidation challenging. Thus, additional study into the dependence of catalytic behavior on the structure of a material (e.g., reactivity observed when specific surface functional groups are present) is warranted. Further exploration of the physical properties of the functionalized carbons (e.g., surface area, pore structure, etc.) may also provide routes to improved catalytic efficiencies as it has been demonstrated that such characteristics can influence the ability of substrates to interact with the carbon surface.7 Ultimately, it is expected that both the structural and electronic properties of a carbocatalyst may be independently tailored to impart regio-, chemo-, or stereoselectivities to various synthetic transformations.
This work was generously supported by the Robert A. Welch Foundation (F-1621).
Notes and references
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- M. S. Dresselhaus, ACS Nano, 2010, 4, 4344–4349 CrossRef CAS.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- D. S. Su, ChemSusChem, 2009, 2, 1009–1020 CrossRef CAS.
- M. Wissler, J. Power Sources, 2006, 156, 142–150 CrossRef CAS.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259–271 CrossRef CAS.
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef CAS.
- R. K. Kalyoncu and H. A. Taylor Jr., in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, Hoboken, NJ, USA, 2005, p. 771 Search PubMed.
- IUPAC, Compendium of Chemical Terminology, Blackwell Scientific, Oxford, 1997 Search PubMed.
- F. C. Cowlard and J. C. Lewis, J. Mater. Sci., 1967, 2, 507–512 CrossRef CAS.
- J. Robertson and E. P. O'Reilly, Phys. Rev. B, 1987, 35, 2946–2957 CrossRef CAS.
- K. Nishimiya, T. Hata, Y. Imamura and S. Ishihara, J. Wood Sci., 1998, 44, 56–61 CrossRef CAS.
- V. Meille, Appl. Catal., A, 2006, 315, 1–17 CrossRef CAS.
- P. Ehrburger, Carbon, 1991, 29, 763–768 CrossRef CAS.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 CAS.
- A. Kutzelnigg, Ber. Dtsch. Chem. Ges. B, 1930, 63, 1753–1758.
- I. M. Kolthoff, J. Am. Chem. Soc., 1932, 54, 4473–4480 CrossRef CAS.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235440 CrossRef.
- D.-S. Yang and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4122–4126 CrossRef CAS.
- A. Bazylak, J. Heinrich, N. Djilali and D. Sinton, J. Power Sources, 2008, 185, 1147–1153 CrossRef CAS.
- F. Ahnert, H. A. Arafat and N. G. Pinto, Adsorption, 2003, 9, 311–319 CrossRef CAS.
- E. K. Rideal and W. M. Wright, J. Chem. Soc. Trans., 1925, 127, 1347–1357 RSC.
- E. K. Rideal and W. M. Wright, J. Chem. Soc., 1926, 129, 1813–1821 Search PubMed.
- E. K. Rideal and W. M. Wright, J. Chem. Soc., 1926, 129, 3182–3190 Search PubMed.
- D. M. Ruthven, N. S. Raghavan and M. M. Hassan, Chem. Eng. Sci., 1986, 41, 1325–1332 CrossRef CAS.
- T. A. Centeno and A. B. Fuertes, Carbon, 2000, 38, 1067–1073 CrossRef CAS.
- G. C. Grunewald and R. S. Drago, J. Am. Chem. Soc., 1991, 113, 1636–1639 CrossRef CAS.
- M. Ai, J. Catal., 1975, 40, 327–333 CrossRef CAS.
- B. H. Davis, J. Catal., 1983, 79, 58–69 CrossRef CAS.
- F. Lücking, H. Köser, M. Jank and A. Ritter, Water Res., 1998, 32, 2607–2614 CrossRef CAS.
- H. J. H. Fenton, J. Chem. Soc. Trans., 1894, 65, 899–910 RSC.
- Y. Kuang, N. M. Islam, Y. Nabae, T. Hayakawa and M.-a. Kakimoto, Angew. Chem., Int. Ed., 2010, 49, 436–440 CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS.
- B. H. Han, D. H. Shin and S. Y. Cho, Tetrahedron Lett., 1985, 26, 6233–6234 CrossRef CAS.
- J. W. Larsen, M. Freund, K. Y. Kim, M. Sidovar and J. L. Stuart, Carbon, 2000, 38, 655–661 CrossRef CAS.
- B. H. Han and D. G. Jang, Tetrahedron Lett., 1990, 31, 1181–1182 CrossRef CAS.
- B. Li and Z. Xu, J. Am. Chem. Soc., 2009, 131, 16380–16382 CrossRef CAS.
- L. Pacosová, C. Kartusch, P. Kukula and J. A. van Bokhoven, ChemCatChem, 2011, 3, 154–156 Search PubMed.
- J. Fu, F. Shi, L. T. Thompson Jr., X. Lu and P. E. Savage, ACS Catal., 2011, 1, 227–231 Search PubMed.
- J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 RSC.
- Y. Suzuki, M. Matsushima and M. Kodomari, Chem. Lett., 1998, 319–320 CrossRef CAS.
- B. Ganem and V. R. Small Jr., J. Org. Chem., 1974, 39, 3728–3730 CrossRef CAS.
- M. Kodomari, Y. Suzuki and K. Yoshida, Chem. Commun., 1997, 1567–1568 RSC.
- G. A. Sereda, V. B. Rajpara and R. L. Slaba, Tetrahedron, 2007, 63, 8351–8357 CrossRef CAS.
- P. G. Klemens and D. F. Pedraza, Carbon, 1994, 32, 735–741 CrossRef CAS.
- B. Garrigues, C. Laporte, R. Laurent, A. Laporterie and J. Dubac, Liebigs Ann., 1996, 739–741 Search PubMed.
- B. Garrigues, R. Laurent, C. Laporte, A. Laporterie and J. Dubac, Liebigs Ann., 1996, 743–744 Search PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS.
- J. Zhang, D. S. Su, R. Blume, R. Schlögl, R. Wang, X. Yang and A. Gajović, Angew. Chem., Int. Ed., 2010, 49, 8640–8644 CrossRef CAS.
- D. S. Su, J. J. Delgado, X. Liu, D. Wang, R. Schlögl, L. Wang, Z. Zhang, Z. Shan and F.-S. Xiao, Chem.–Asian J., 2009, 4, 1108–1113 CrossRef CAS.
- N. Keller, N. I. Maksimova, V. V. Raddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlögl, Angew. Chem., Int. Ed., 2002, 41, 1885–1888 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Catal., 2002, 209, 35–42 CrossRef CAS.
- G. I. Panov, K. A. Dubkova and E. V. Starokon, Catal. Today, 2006, 117, 148–155 CrossRef CAS.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281–288 CrossRef CAS.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- S. Cerveny, F. Barroso-Bujans, Á. Alegría and J. Colmenero, J. Phys. Chem. C, 2010, 114, 2604–2612 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- A. Lerf, H. He, T. Riedl, M. Forster and J. Klinowski, Solid State Ionics, 1997, 101–103, 857–862 CrossRef CAS.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- B. V. Lebedev, L. Y. Tsvetkova and K. B. Zhogova, Thermochim. Acta, 1997, 299, 127–131 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2008, 4, 25–29.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang and H. Liu, J. Phys. Chem. C, 2008, 112, 8192–8195 CrossRef CAS.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Gao, B. Liu, C. Jiang and H.-M. Cheng, Carbon, 2009, 47, 493–499 CrossRef CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- H. P. Boehm, A. Clauss, G. O. Fischer and U. Hofmann, Z. Anorg. Allg. Chem., 1962, 316, 119–127 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- J. Pyun, Angew. Chem., Int. Ed., 2011, 50, 46–48 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443–3447 RSC.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011 DOI:10.1016/j.tet.2011.1002.1065.
- S. Astin, L.de V. Moulds and H. L. Riley, J. Chem. Soc., 1935, 901–904 RSC.
- V. Nair, L. G. Nair, S. B. Panicker, V. Sheeba and A. Augustine, Chem. Lett., 2000, 584–585 CrossRef CAS.
- H. Firouzabadi, A. R. Sardarian, H. Moosavipour and G. M. Afshari, Synthesis, 1986, 285–288 CrossRef CAS.
- J. F. Froning and G. F. Hennion, J. Am. Chem. Soc., 1940, 62, 653–655 CrossRef CAS.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Adv. Synth. Catal., 2011, 53, 528–532 CrossRef.
- A. Hamwi and V. Marchand, J. Phys. Chem. Solids, 1996, 57, 867–872 CrossRef CAS.
- C.-L. Sun, H. Li, Da-Gang Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS.
- N. E. Leadbeater and M. Marco, J. Org. Chem., 2003, 68, 5660–5667 CrossRef CAS.
- N. E. Leadbeater, M. Marco and B. J. Tominack, Org. Lett., 2003, 5, 3919–3922 CrossRef CAS.
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS.
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS.
- S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586–5587 CrossRef CAS.
- N. E. Leadbeater, Nat. Chem., 2010, 2, 1007–1009 CrossRef CAS.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef CAS.
- D. Piasta, C. Bellmann, S. Spange and F. Simon, Langmuir, 2009, 25, 9071–9077 CrossRef CAS.
- D. G. Hager, Environ. Sci. Technol., 1967, 1, 287–291 CrossRef CAS.
- X. D. Dai, X. M. Liu, L. Qian, K. Qiao and Z. F. Yan, Energy Fuels, 2008, 22, 3420–3423 CrossRef CAS.
- H. Sharghi and M. H. Sarvari, Synthesis, 2003, 243–246 CrossRef CAS.
- J. Marquié, A. Laporterie, J. Dubac and N. Roques, Synlett, 2001, 493–496 CAS.
- H. B. Kagan, Pure Appl. Chem., 1976, 46, 177–181 CrossRef CAS.
Footnote |
| † Although the authors of these studies attributed this behavior to the presence of acidic species adsorbed onto the charcoal surface, the use of strongly acidic and oxidizing environments may have introduced covalently bound acidic groups directly to the surface, as is seen in the preparation of graphite oxide (GO) and other similar materials.55 |
| This journal is © The Royal Society of Chemistry 2011 |