Hydrogenation in flow: Homogeneous and heterogeneous catalysis using Teflon AF-2400 to effect gas–liquid contact at elevated pressure†‡
Matthew
O'Brien
,
Nicholas
Taylor
,
Anastasios
Polyzos
,
Ian R.
Baxendale
and
Steven V.
Ley
*
Whiffen laboratory, University Chemical Laboratories, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk
First published on 1st April 2011
Abstract
A Tube-in-Tube reactor/injector has been developed, based on a gas-permeable Teflon AF-2400 membrane, which allows both heterogeneous and homogeneous catalytic hydrogenation reactions to be efficiently carried out at elevated pressure in flow, thereby increasing the safety profile of these reactions. Measurements of the gas permeation through the tubing and uptake into solution, using both a burette method and a novel computer-assisted ‘bubble counting’ technique, indicate that permeation/dissolution follows Henry's law and that saturation is achieved extremely rapidly. The same gas-permeable membrane has also been shown to efficiently effect removal of excess unreacted hydrogen, thus enabling further downstream reaction/processing.
Introduction
Since the initial ground-breaking work by Sabatier and Senderens over a century ago,1 the catalytic hydrogenation of multiple bonds has become one of the most widely used and important reactions in organic synthesis. It is the cornerstone of many processes which are of huge economic and social importance, and research into the reaction continues unabated as new applications emerge.2There are many factors contributing to the popularity of hydrogenation as a chemical process. From an economic standpoint, hydrogen is very inexpensive and readily available.3 As it is a gas, it can be used in excess, to drive reactions to completion in a timely manner, with the unreacted gas being easily removed at the end of the process. It is also relatively non-toxic.
However, there are some very serious drawbacks to the use of hydrogen that must be taken into consideration. As all chemists will be aware from classroom demonstrations, it forms extremely flammable and explosive mixtures with oxygen (and therefore air). Indeed, Henry Cavendish, one of the early pioneers of hydrogen chemistry, gave it the name ‘inflammable air’.4 Perhaps due to the tragic circumstances (and images thereof) in which the dirigible LZ 129 Hindenburg was destroyed at the Lakehurst naval station in 1937, the hazardous nature of hydrogen gas has also been firmly engrained in the public consciousness.
In fact, this hazard is greatly compounded by the gaseous nature of hydrogen. Many hydrogenation reactions require high concentrations of hydrogen in order to proceed at an acceptable rate and, of course, this concentration is approximately proportional to pressure (Boyle's law for gas-phase, Henry's law for solutions). So, obtaining high concentrations necessitates using high pressures and this is a serious safety consideration often requiring substantial technical and economic investment to ensure safe operation. Unfortunately, despite efforts to maximise safety, there have been several major explosions at hydrogenation facilities, some tragically involving loss of life.5
The potential energy stored in a pressurised container (and therefore released in the event of an accidental breakdown) is approximately proportional to its volume. This is also true of the chemical energy released in the event of an accidental ignition. These considerations suggest that, for the hydrogenation of a fixed amount of material, the adoption of a continuous processing strategy that uses a low operating volume reactor would be highly desirable from a safety viewpoint when compared with the corresponding higher volume batch process.
Flow chemistry has emerged over recent years as an enabling technology that can provide an enhanced safety profile for reactions that involve hazardous or explosive intermediates, or for processes which involve high temperatures or pressures, and additionally often facilitates rapid reactions involving short-lived reactive intermediates.6 For gas–liquid contacting in flow, typical reactor designs have involved plug flow, or mechanical mixing of the two phases.7 Seeking a more efficient, controllable and reliable method of gas–liquid contact, research in our laboratory has been focused on the use of semi-permeable membranes, which provide a very high effective surface area and allow gas (but not liquids) to pass from one side to the other. We recently developed a continuous-flow ozonolysis process that allowed for the quenching of potentially explosive ozonides (and peroxy compounds) as they were formed, incorporating a Teflon AF-2400 membrane to facilitate permeation of the O2/O3 gas mixture into the substrate flow stream.8 We subsequently developed a ‘Tube-in-Tube’ configuration, which we used to carry out a series of carboxylation reactions.9 In this paper we wish to report the extension of this concept to the important problem of homogeneous and heterogeneous catalytic hydrogenation reaction at elevated pressure. We also report our initial findings on the measurement of gas transfer through the membrane into solution and its relationship to pressure and flow rate.
Tube-in-Tube gas–liquid flow reactor
The basic reactor apparatus10 used in this study is shown in Fig. 1 (Caution!! This reactor is a prototype. It has not been fully tested for safety. All experiments using hydrogen under pressure should be carried out only with adequate ventilation and fire prevention facilities in place which are capable of handling a sudden mechanical failure and auto-ignition). The solution (dyed red in Fig. 1 for clarity) enters the apparatus via the connector at point (a). At point (b), the solution passes through a T-piece and into the inner Teflon AF-2400 tube11 (0.8 mm o.d., 0.6 mm i.d., 0.1 mm wall thickness) of the Tube-in-Tube gas–liquid contactor. A gas line (c), which comes from a regulator on the hydrogen cylinder, is connected, via T-piece (b), to the outer PTFE tube (3.18 mm o.d., 1.59 mm i.d., 0.80 mm wall thickness) of the Tube-in-Tube contactor. It is in this Tube-in-Tube contactor that gas permeates into the solution which is being pumped through the inner tube (see left inset for close-up of Tube-in-Tube). This material was chosen for the outer tube for reasons of convenience as it is both translucent (thereby allowing visual inspection of the contents) and flexible. Steel tubing could also have been used instead (it is possible that some slight leakage of hydrogen might occur through the outer PTFE tubing). Only the inner tube of the Tube-in-Tube reactor is made of Teflon AF-2400, at all other times the solution passes through standard PFA tubing. The hydrogenated solution passes out of the reactor via T-piece (g) through the back-pressure regulator (k). Hydrogen pressure is measured using the gauge (e). The pressure can be manually released via the needle valve (j). The safety release valve (l) is adjustable and allows the controlled release of pressure if this exceeds the desired threshold, in this case 60 bar. The presence of the back-pressure regulator is very important for the correct operation of the reactor as it creates a pressure drop and thereby helps to keep the gas in solution. Without it, outgassing of hydrogen occurs causing erratic flow. At all times during the course of this work, the solution of hydrogen and reactants was homogeneous, no bubbles being observed in the line until after the solution passes through the back-pressure regulator where pressure is released. Teflon AF-2400, which is a copolymer of tetrafluoroethylene and perfluorodimethyldioxolane, is an ideal choice of material for the purposes of gas–liquid contact. It combines extremely high levels of gas permeability for a wide variety of gases with practically zero liquid permeability and a broad chemical resistance commonly associated with perfluorinated polymers.12 | ||
| Fig. 1 Hydrogenation reactor/injector. Key: a) solution inlet; b) Swagelok T-piece; c) gas inlet (1/8′′ o.d. PTFE tubing); d) Tube-in-Tube gas–liquid contactor (inner tube is 0.8 mm o.d. Teflon AF-2400, outer tube is 1/8′′ o.d. PTFE); e) pressure gauge; f), g), h) Swagelok T-pieces; i) 1/8′′ stainless steel connector between T-pieces g and h; j) Swagelok needle valve; k) solution outlet (connected to back-pressure regulator); l) pressure relief valve (set to release pressure at 60 bar). Left inset: close-up view of the Tube-in-Tube configuration, solution dyed red. Right inset: simplified diagrammatical view of connections at T-piece b. | ||
Homogeneous hydrogenation
To investigate the potential of the reactor in homogeneous hydrogenation reactions we began with the hydrogenation of ethyl cinnamate 1 using Crabtree's catalyst 2 (0.001 equiv.) in dichloromethane.13 The general apparatus setup is outlined in Fig. 2. Using a Uniqsis Flowsyn,14 the substrate 1 (0.5 M, DCM) and catalyst 2 (0.0005 M, DCM) were introduced via injection loops (2 mL and 3 mL respectively) into two separate streams. The injection loop for the catalyst was longer than that of the substrate and injection of the catalyst was timed to begin slightly in advance of the injection of the reagent, thus ensuring that the substrate would not enter the reactor without the catalyst also being present. The two solution streams were united at a T-piece before entering the reactor. The length of the Tube-in-Tube gas contacting section was 100 cm. In the initial experiment, no additional residence loop was used, although this was investigated subsequently. A 250 psi (17.2 bar) back-pressure regulator ensured that the upstream hydrogen remained in solution until the pressure was released after the back-pressure regulator. All reactions were carried out at ambient temperature. The valve connecting the hydrogen regulator to the reactor was opened before the reactant streams were injected. The dissolved hydrogen could be observed outgassing from the solution as it emerged from the back-pressure regulator. At a combined flow rate of 2 mL min−1 (corresponding to a residence time in the Tube-in-Tube reactor of approximately 8 s), and 20 bar of hydrogen (relative to ambient) the conversion to product 3 (as measured by 1H NMR analysis) was 48%. | ||
| Fig. 2 Schematic of homogeneous flow hydrogenation apparatus. | ||
Although the amount of outgassing from the product stream was noticeably diminished for a period during the reaction, it was clear not all the hydrogen was being consumed by the reaction. It therefore seemed reasonable that more conversion might be achieved if the reaction time was extended. To investigate this, we placed various additional residence loops between the reactor/injector and the back-pressure-regulator. The results of this are shown in Fig. 3.
 | ||
| Fig. 3 Dependence of the conversion of 1 to 3 on total residence time in reactor and additional residence loop. aConversion based on 1H NMR analysis. | ||
As can be seen, at low volume (and therefore low residence time) the conversion is strongly dependent on the volume of the additional loop. However, the conversion plateaued just over 70% at higher volumes. Interestingly, with the 5 mL residence loop (158.5 s residence time), outgassing was no longer observed after the back-pressure regulator, indicating that most likely all the hydrogen had been consumed. With hydrogen now seemingly the limiting factor, we turned our attention to the variation of conversion with gas pressure, keeping a constant total flow rate of 2 mL min−1 and using the 2 mL residence loop (68.5 s residence time). The results are shown in Fig. 4.
 | ||
| Fig. 4 Dependence of the conversion of 1 to 3 on hydrogen pressure. aConversion based on 1H NMR analysis. | ||
Unsurprisingly, the conversion was highly pressure dependent. Indeed, when the reaction was carried out under 30 bar of hydrogen, complete conversion to product 3 was achieved. With these results in hand, we then carried out the hydrogenation on a series of other alkenes. A pressure of 25 bar was used. To ensure complete conversion, a slightly lower combined flow rate (1.4 mL min−1) and 0.002 equiv of catalyst were used (0.003 equiv taking into account the surplus catalyst in the longer injection loop); the results are shown in Table 1. All products were isolated in quantitative yield after removal of solvent and in high purity (by NMR spectroscopy; residual catalyst could not be observed at the very low levels used). Given that the combined residence time in the reactor/injector and downstream residence loop is around 93 s, these reactions are extremely rapid.




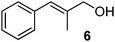

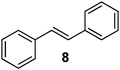






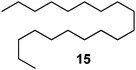

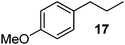
Measurement of hydrogen dissolution (via outgassing)
Although we had shown that the conversion of the hydrogenation was dependent on both residence time and pressure, we sought to obtain a quantitative analysis of the gas permeation and solvation processes taking place in the reactor. To make this generally applicable to a range of processes, we decided to measure gas uptake in pure DCM alone, in the absence of reactants/catalysts which could provide a source of variability. The direct measurement of the amount of gas being taken up, whilst possible, would be technically very challenging. Instead, we decided to measure the amount of gas venting from the solution downstream of the back-pressure regulator, when the total pressure of solution could relax to atmospheric pressure. Assuming that the residual gas remaining dissolved in the solvent was small (owing to the low solubility of hydrogen in DCM at atmospheric pressure)15 relative to total hydrogen dissolved at pressure, measuring the volume of gas released would be a reasonably accurate measure of the amount of gas being taken up by the flow stream in the reactor. As well as successfully using a traditional burette method, we have also investigated the use of a novel computer based ‘bubble counting’ approach.Using a typical burette setup with a graduated tube filled with DCM and suspended upright in a beaker of DCM, the time taken to collect a fixed volume of gas was measured at different applied pressures of hydrogen (5, 7.5 and 10 bar) for a series of flow rates (the volume of gas measured was such that the inner and outer solvent levels were at the same height at the moment of measurement). The volume of hydrogen outgassed per second divided by the volume of DCM per second (the pumping flow rate) was then calculated as a measure of hydrogen concentration above that at atmospheric pressure. For these gas measurements, a shorter 50 cm length of Tube-in-Tube gas contactor was used, to increase the likelihood that the solution would not be saturated over the entire range of flow rates used (in which case the plot of hydrogen against flow rate would simply be a flat line). The results of the burette measurements are shown in Fig. 5. At short residence times (high flow rates), the concentration of hydrogen varies approximately linearly with residence time. At longer residence times the concentration of hydrogen appears to reach a plateau, suggesting that it has reached saturation. Taking the flattened portion of the line as the saturation concentration, the system approximately appears to obey Henry's law. It is of note that the solution appears to reach saturation after only 5–10 s, indicating a very high effective surface area to volume ratio, enabling rapid diffusion and therefore high rates of reaction.
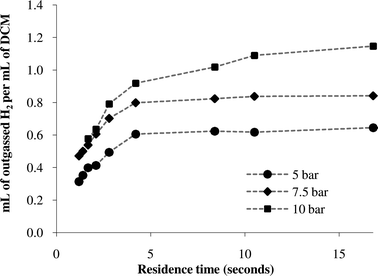 | ||
| Fig. 5 Burette measurements of hydrogen outgassing. | ||
For a thorough analysis of permeation/solvation over a broader range of solvents, flow rates and pressures, it would seem highly desirable to move towards a fully automated process. Seeking an alternative to the burette measurement, whose labour intensive operation might be technically complicated to automate, we envisaged a computer-assisted approach of ‘bubble counting’. A length of tubing connected to the solvent outlet (downstream of the back-pressure regulator) was wrapped many times around a rectangular support which was held in place in the view of a digital camera. Digital images of this tube section, which contained plugs of outgassed hydrogen, were taken and then digitally processed by a computer to calculate the total volume of the plugs. To facilitate this processing, we added a small amount of red dye (Sudan red 7B) to the DCM. In this way, the processing was essentially reduced to counting the number of red pixels in the photograph. This was easily achieved using a simple computer programme written in Python16 (see supporting info.†). Shown in Fig. 6 are two photographs of the wrapped tubing.
 | ||
| Fig. 6 Bubble-counting method a) Calibration run, reactor not pressurised b) corresponding converted image with pixels only pure red or pure white c) photograph of outlet tubing from pressurised reactor d) converted image from pressurised reactor. | ||
Image (a) is a calibration run with no outgassed hydrogen and image (c) was taken after pressurising the reactor. Also shown are two converted images, (b) and (d), used to visually check the counting programme, where each pixel has been replaced with either a pure red or pure white pixel according to the decision making algorithm of the programme. The approximate concentration of hydrogen was calculated as the number of extra white pixels not present in the calibration photograph (volume of gas) divided by the number of red pixels (volume of liquid).
The results from a series of runs at different pressures and flow rates are shown in Fig. 7. As can be seen the results match well to those from the burette measurements including the general shapes of the plotted curves.
 | ||
| Fig. 7 Digital bubble-counting measurements of hydrogen outgassing. | ||
As it is possible to place the camera, the pumping system and the gas pressure regulator under computer control, we envisage the incorporation of this technique into a fully automated system. This digital pixel counting method could facilitate the measurement of both kinetic permeation rates and equilibrium (solubility at saturation) gas solubility data for a wide range of gases and liquids in a way which is technically less demanding and more rapid than many currently available methods.17 We are currently working to implement these solutions into our other flow chemistry platforms.
Gas removal
One of the key advantages of flow chemistry is that several processes can be performed in multi-step sequences by passing the reactant stream through a series of reactors and/or purification cartridges.18 For gas-liquid reactions to be used in these continuous multi-step processes, it would be desirable to be able to remove the excess gas present in the reactant stream prior to entering any downstream process, thus ensuring complete chemical and mechanical compatibility with downstream operations. It seemed plausible that this could be achieved simply by passing the reactant stream through a second gas-exchanger which was not pressurised (or even under vacuum). To investigate this possibility, the outlet from the reactor was passed, via modified plastic ‘quick-fit’ stoppers through a glass vessel before continuing to the pixel-counting apparatus, as shown schematically in Fig. 8. | ||
| Fig. 8 Schematic of gas-remover and bubble counting apparatus. | ||
This degassing device had an outlet valve which could be opened to the atmosphere or connected to a vacuum pump. Inside the device, the solution passed through a 45 cm length of Teflon AF-2400. Initially, the reactor was pressurised to 5 bar of H2 and the back-pressure regulator was placed after the main reactor but before this gas-exchanger whose outlet was open to the atmosphere. As shown in Fig. 9b, this had only a small effect on the observed outgassed hydrogen (0.45 mL/mL) compared with the amount measured when the gas-exchanger was bypassed (0.59 mL/mL), Fig. 9a. When the back-pressure regulator was placed after the gas-exchanger, the measured outgassed hydrogen was significantly reduced (0.06 mL/mL), Fig. 9c. It seems that if the solution is allowed to depressurise to atmospheric pressure, the concentration, or partial pressure, of hydrogen inside the tube will be effectively low, and this retards its permeation through the Teflon AF-2400. If the solution is kept at a higher pressure (by the use of a back-pressure regulator), the concentration, or partial pressure, of the hydrogen remains high, facilitating its permeation. By placing the gas-exchanger vessel under vacuum (ca. 1 mm Hg), the measured outgassing was reduced to zero, indicating that practically all the gas had been removed in the gas-removal vessel (Fig. 9d). This result opens up the possibility of performing continuous multi-step (and even multi-gas) reaction sequences.
 | ||
| Fig. 9 Photographs of outgassed hydrogen in bubble counting array with or without gas-remover (50 cm Tube-in-Tube reactor pressurised to 5 bar H2). a) no degassing vessel b) back-pressure regulator before the degassing vessel c) back-pressure regulator after the degassing vessel d) vacuum applied to the degassing vessel. | ||
Heterogeneous catalytic hydrogenation
Having established that this system is effective for homogeneous hydrogenation, we sought to investigate its use in heterogeneous hydrogenation. For this purpose we used a similar reaction setup (Fig. 10), but with an omnifit glass column (packed with 250 mg of 10% palladium on carbon catalyst, 7.7 mol% wrt substrate) placed between the Tube-in-Tube reactor/injector and the back-pressure regulator. Initially we chose to study the hydrogenation of ethyl cinnamate 1 in ethyl acetate on a 3.0 mmol scale. | ||
| Fig. 10 Schematic of heterogeneous hydrogenation apparatus. | ||
We began by investigating the variation of conversion with pressure, in single-pass runs. The plotted results, shown in Fig. 11, reveal that there is a strong, approximately linear, pressure dependence in the conversion range covered. For safety reasons (bearing in mind that the reactor was a prototype and not fully tested), we did not want to simply increase the pressure of hydrogen and, rather than increase the catalyst loading, we chose to investigate the option of recycling the product back through the reactor.
 | ||
| Fig. 11 Variation of conversion (of 2 to 3) with pressure for single-pass heterogeneous hydrogenation. aConversion measured by 1H NMR. | ||
This was achieved by simply placing the pump inlet lines into the product collection flask. The reaction was performed on a 5.0 mmol scale with the same amount of catalyst under 15 bar of hydrogen pressure and progress was monitored by TLC and 1H NMR analysis of small aliquots. Conversion was complete after 290 min at which point the product was flushed from the system (by pumping through fresh ethyl acetate) and isolated in quantitative yield after removal of solvent. A series of alkenes were then hydrogenated using this recycling configuration, on a 5.0 mmol scale under 15 bar of hydrogen with a larger cartridge of catalyst (750 mg, 0.71 mmol Pd, 14% wrt substrate), see Table 2. The same cartridge of catalyst was used for all the reactions, each of which reached completion in around 2 h (except for the alkyne which took approximately twice as long, entry 6). All products were isolated in quantitative yield and high purity (by 1H and 13C NMR analysis). Assuming a molar volume of 22.414 L mol−1 at atmospheric pressure, the 5.0 mmol of hydrogen consumed equates to 112 mL (10.0 mmol/ 224 mL for entry 6). To investigate the ease of scale-up, the hydrogenation of ethyl cinnamate 1 was repeated on a 10 mL (10.49 g, 60 mmol) scale. In this case, in order to facilitate rapid conversion to the product, a higher pressure (25 bar) of hydrogen was used, along with a higher flow rate (3 mL min−1). The same cartridge of catalyst was used (750 mg, 0.71 mmol, 1% wrt substrate). Outgassing, which was lost 3 min after commencing the reaction, returned after 6 h, indicating reaction completion. After removal of the solvent, the product was isolated in quantitative yield. The 60 mmol of hydrogen consumed in this example equates to 1.34 L (at atmospheric pressure).
| Entry | Substrate | Product | Timea | Yieldb |
|---|---|---|---|---|
| a Time (in minutes) for complete conversion as monitored by return of outgassing and/or TLC/NMR analysis, reactions performed on 5 mmol unless otherwise stated. b Isolated yield after removal of solvent. c Reaction performed on a 60 mmol scale (10.49g). | ||||
| 1 |

|

|
125 | Quant. |
| 360c | Quant. | |||
| 2 |

|
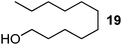
|
130 | Quant. |
| 3 |
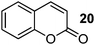
|

|
120 | Quant. |
| 4 |
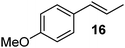
|
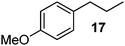
|
115 | Quant. |
| 5 |

|

|
120 | Quant. |
| 6 |

|

|
250 | Quant. |
| 7 |
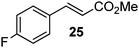
|

|
108 | Quant. |
| 8 |
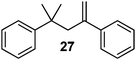
|
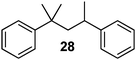
|
110 | Quant. |
Conclusions
From this work we have established an effective, economic and scalable Tube-in-Tube reactor/injector for both homogeneous and heterogeneous catalytic hydrogenation, whereby only a minimal gas volume required pressurisation therefore improving the safety profile of the system. The gas is efficiently and rapidly delivered to the reacting flow stream via permeation through a central semipermeable Teflon AF-2400 tube. Furthermore, we have shown both with a simple burette system and a novel computer-assisted bubble counting technique the levels of hydrogen uptake in the device. The bubble counting technique has the potential to facilitate the rapid and efficient computer-assisted measurement of gas solubility in liquids in general. This will be very useful in our ongoing investigations of flow chemistry using other reactive gases (e.g. CO, Cl2, acetylene, ethylene, SO2, O2etc.) and may find application beyond the scope of this research. Importantly, we have also shown that the same gas-permeable membrane can be used to effect removal of excess unreacted hydrogen thereby providing an opportunity for further machine coupling to effect multi-step flow chemistry processing.Notes and references
- (a) P. Sabatier and J. B. Senderens, C.R. Hebd. Seances Acad. Sci., 1900, 131, 40–42 Search PubMed; (b) P. Sabatier and J. B. Senderens, C. R. Hebd. Seances Acad. Sci., 1905, 140, 482–486 Search PubMed; (c) P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- (a) R. Noyori, T. Ohkuma, M. Kitamura, H. Takaya, N. Sayo, H. Kumobayashi and S. Akutagawa, J. Am. Chem. Soc., 1987, 109, 5856–5858 CrossRef CAS; (b) T. P. Dang and H. B. Kagan, J. Chem. Soc. D, 1971, 481 RSC; (c) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106–112 CrossRef CAS; (d) H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS; (e) R. H. Crabtree and M. W. Davis, J. Org. Chem., 1986, 51, 2655–2661 CrossRef CAS; (f) J. M. Brown, Angew. Chem., Int. Ed. Engl., 1987, 26, 190–203 CrossRef; (g) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC; (h) S. Nishimura, Handbook of heterogeneous catalytic hydrogenation for organic synthesis, J. Wiley, New York, 2001 Search PubMed; (i) J. G. de Vries and C. J. Elsevier, The handbook of homogeneous hydrogenation, Wiley-VCH, Weinheim, 2006 Search PubMed; (j) A. Pfaltz, J. Blankenstein, R. Hilgraf, E. Hörmann, S. McIntyre, F. Menges, M. Schönleber, S. P. Smidt, B. Wüstenberg and N. Zimmermann, Adv. Synth. Catal., 2003, 345, 33–43 CrossRef CAS.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- H. Cavendish, Philos. Trans. R. Soc. London, 1766, 56, 141–184 CrossRef.
- (a) G. Eigenberger and U. Wegerle, ACS Symp. Ser., 1982, 196, 133–143 CrossRef CAS; (b) J. A. MacDiarmid and G. J. T. North, Plant/Oper. Prog., 1989, 8, 96–99 Search PubMed; (c) F. Dryer, M. Chaos, Z. Zhao, J. Stein, J. Alpert and C. Homer, Combust. Sci. Technol., 2007, 179, 663–694 CrossRef CAS; (d) B. J. Lee and I.-S. Jeung, Int. J. Hydrogen Energy, 2009, 34, 8763–8769 CrossRef CAS.
- (a) J. C. Brandt and T. Wirth, Beilstein J. Org. Chem., 2009, 5, 30 Search PubMed; (b) G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708–5723 CrossRef CAS; (c) T. Razzaq and C. O. Kappe, Chem. Asian J., 2010, 5, 1274–1289 CAS; (d) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (e) C. Wiles and P. Watts, Eur. J. Org. Chem., 2008, 1655–1671 CrossRef CAS; (f) M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577–1586 RSC; (g) C. D. Smith, I. R. Baxendale, S. Lanners, J. J. Hayward, S. C. Smith and S. V. Ley, Org. Biomol. Chem., 2007, 5, 1559–1561 RSC; (h) I. R. Baxendale, J. J. Hayward and S. V. Ley, Comb. Chem. High Throughput Screening, 2007, 10, 802–836 CrossRef CAS; (i) M. Baumann, I. R. Baxendale and S. V. Ley, Molecular Diversity, DOI:10.1007/s11030-010-9282-1; (j) I. R. Baxendale and S. V. Ley, Chimia, 2008, 62, 162–168 CrossRef CAS; (k) V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS; (l) B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS; (m) M. Brasholz, J. M. Macdonald, S. Saubern, J. H. Ryan and A. B. Holmes, Chem.–Eur. J., 2010, 16, 11471–11480 CrossRef CAS; (n) A. R. Bogdan and N. W. Sach, Adv. Synth. Catal., 2009, 351, 849–854 CrossRef CAS; (o) A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547–8550 CrossRef CAS; (p) P. J. Nieuwland, K. Koch, N. van Harskamp, R. Wehrens, J. C. M. van Hest and F. P. J. T. Rutjes, Chem.–Asian J., 2010, 5, 799–805 CrossRef CAS; (q) J.-I. Yoshida, Chem. Rec., 2010, 10, 332–341 CrossRef CAS.
- (a) C. Csajagi, B. Borcsek, K. Niesz, I. Kovacs, Z. Szekelyhidi, Z. Bajko, L. Urge and F. Darvas, Org. Lett., 2008, 10, 1589–1592 CrossRef CAS; (b) R. V. Jones, L. Godorhazy, N. Varga, D. Szalay, L. Urge and F. Darvas, J. Comb. Chem., 2006, 8, 110–116 CrossRef CAS; (c) T. Fukuyama, T. Rahman, N. Kamata and I. Ryu, Beilstein J. Org. Chem., 2009, 5, 34 Search PubMed; (d) M. T. Rahman, T. Fukuyama, N. Kamata, M. Sato and I. Ryu, Chem. Commun., 2006, 2236–2238 RSC; (e) P. W. Miller, L. E. Jennings, A. J. deMello, A. D. Gee, N. J. Long and R. Vilar, Adv. Synth. Catal., 2009, 351, 3260–3268 CrossRef CAS; (f) Y. Wada, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2006, 45, 8036–8042 CrossRef CAS; (g) R. D. Chambers, M. A. Fox, G. Sandford, J. Trmcic and A. Goeta, J. Fluorine Chem., 2007, 128, 29–33 CrossRef CAS; (h) S. Hübner, U. Bentrup, U. Budde, K. Lovis, T. Dietrich, A. Freitag, L. Küpper and K. Jähnisch, Org. Process Res. Dev., 2009, 13, 952–960 Search PubMed; (i) S. Saaby, K. R. Knudsen, M. Ladlow and S. V. Ley, Chem. Commun., 2005, 2909–2911 RSC; (j) M. Baumann, I. R. Baxendale and S. V. Ley, Synlett, 2010, 749–752 CAS; (k) V. Franckevicius, K. R. Knudsen, M. Ladlow, D. A. Longbottom and S. V. Ley, Synlett, 2006, 889–892 CrossRef CAS; (l) C. F. Carter, I. R. Baxendale, M. O'Brien, J. B. J. Pavey and S. V. Ley, Org. Biomol. Chem., 2009, 7, 4594–4597 RSC; (m) R. Abdallah, V. Meille, J. Shaw, D. Wenn and C. de Bellefon, Chem. Commun., 2004, 372–373 RSC; (n) E. V. Rebrov, M. J. F. Warnier, O. Muraza, M. H. J. M. De Croon, V. Hessel and J. C. Schouten, Proceedings of the AIChE, Houston Texas, USA, 2007 Search PubMed; (o) E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS.
- M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596–1598 CrossRef CAS.
- A. Polyzos, M. O'Brien, T. P. Petersen, I. R. Baxendale and S. V. Ley, Angew. Chem., Int. Ed., 2011, 50, 1190–1193 CrossRef CAS.
- Manifold manufactured by Cambridge Reactor Design Ltd, Twentypence Road, Cottenham, Cambridgeshire, CB24 8PS, UK. http://www.cambridgereactordesign.com.
- Teflon AF-2400 tubing was supplied by Biogeneral Inc., 9925 Mesa Rim Road, San Diego, California, USA. http://www.biogeneral.com.
- (a) P. R. Resnick, US Patent 3978030; (b) P. R. Resnick and W. H. Buck, in Fluoropolymers II, ed. G. Hougham, P. E. Cassidy, K. Johns and T. Davidson, Kluwer Academic, New York, pp. 25–33, 1999 Search PubMed; (c) S. M. Nemser and I. C. Roman, US Patent 5051114; (d) I. Pinnau and L. G. Toy, J. Membr. Sci., 1996, 109, 125–133 CrossRef CAS.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- Uniqsis Ltd, 29 Station Road, Shepreth, Cambridgeshire, UK, SG8 6GB. http://www.uniqsis.com.
- K. Shirono, T. Morimatsu and F. Takemura, J. Chem. Eng. Data, 2008, 53, 1867–1871 CrossRef CAS.
- http://www.python.org/ .
- (a) J. Tan, J. Xu, K. Wang and G. Luo, Ind. Eng. Chem. Res., 2010, 49, 10040–10045 CrossRef CAS; (b) M. Doan Cong and M. Ruel, J. Chem. Eng. Data, 1973, 18, 41–44 CrossRef; (c) R. Battino and H. L. Clever, Chem. Rev., 1966, 66, 395–463 CrossRef CAS; (d) L. H. Turner, Y. C. Chiew, R. C. Ahlert and D. S. Kosson, AIChE J., 1996, 42, 1772–1788 CrossRef CAS.
- (a) S. V. Ley, A. Massi, F. Rodriguez, D. C. Horwell, R. A. Lewthwaite, M. C. Pritchard and A. M. Reid, Angew. Chem., Int. Ed., 2001, 40, 1053–1055 CrossRef CAS; (b) I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566–2568 RSC; (c) I. R. Baxendale and S. V. Ley, Ernst Schering Found. Symp. Proc., 2006, 151–185 Search PubMed; (d) S. V. Ley, C. Mitchell, D. Pears, C. Ramarao, J. Q. Yu and W. Zhou, Org. Lett., 2003, 5, 4665–4668 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of products, further experimental detail. See DOI: 10.1039/c1sc00055a |
| ‡ We acknowledge funding from EPSRC (MOB), CSIRO (AP), the BP 1702 professorship (SVL) and the Royal Society (IRB). |
| This journal is © The Royal Society of Chemistry 2011 |