Enantioselective synthesis of cyclic carbamimidates via a three-component reaction of imines, terminal alkynes, and p-toluenesulfonylisocyanate using a monophosphine gold(I) catalyst†
Matthew J.
Campbell
and
F. Dean
Toste
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@berkeley.edu; Fax: +510-666-2504; Tel: +510-643-3049
First published on 12th May 2011
Abstract
A racemic Au(I)-catalyzed three-component reaction has been developed to prepare cyclic carbamimidates from imines, terminal alkynes, and sulfonylisocyanates. This reaction exploits the carbophilic π-acidity of gold catalysts to first activate an alkyne toward deprotonation and secondly, to activate the internal alkyne generated toward intramolecular O-cyclization. Unlike similar previously reported multicomponent gold-catalyzed reactions, the stereocenter generated during the alkynylation is preserved in the product. This trait was exploited by developing an enantioselective variant, using an unusual trans-1-diphenylphosphino-2-arylsulfamidocyclohexane ligand. Moderate to excellent levels of enantioselectivity were obtained using a variety of N-arylbenzylidene anilines (41–95% ee, 18 examples).
Introduction
Multicomponent one-pot transformations that directly provide complex products are of synthetic interest due to their innate efficiency, simplicity, and elegance. Classic three-component reactions such as the Mannich,1 Ugi,2 Biginelli,3 and Passerini4 reactions have been used extensively in modern organic synthesis, especially within combinatorial chemistry.5 The development of novel multicomponent reactions remains a high priority, and progress has been brisk despite the challenges inherent in controlling the individual reaction rates and pre-equilibria of the various components.Gold(I) and gold(III) complexes have recently been utilized to catalyze a wide variety of transformations.6 Specifically, gold complexes have been employed to catalyze one or more of the individual transformations in three-component reactions, such as the activation and addition of alkynes to iminium ions,7 addition or cyclization of carbon or heteroatom nucleophiles onto alkynes,7e, 7h, 8 the oxidation of susceptible amines (dihydroquinolines), as well as the newly disclosed alkoxy- and hydroxyarylation reactions between alcohols or water, olefins, and arylboronic acids.9 Notably, to our knowledge, no asymmetric multicomponent reactions have been disclosed in which a chiral gold complex controls a facially selective alkynylation of imines.10 We set a goal to close this technology gap while exploiting the remarkable properties of Au(I) salts (carbophilic π-acidity, functional group tolerance, and stability towards water and oxygen).
We were able to draw upon several seminal reports of gold-catalyzed three-component reactions as a starting point for reaction development. In particular, we were intrigued by reports detailing the ability of gold salts to first activate terminal alkynes for the addition to iminium ions and then promote the intramolecular cyclization of suitable nucleophiles onto the internal alkyne-gold π-complex. However, we wished to evaluate new processes because two of the three known multicomponent transformations proceeded with destruction of the newly formed propargylamine stereocenter, which we wished to set (and subsequently preserve) through an asymmetric alkynylation (Scheme 1).
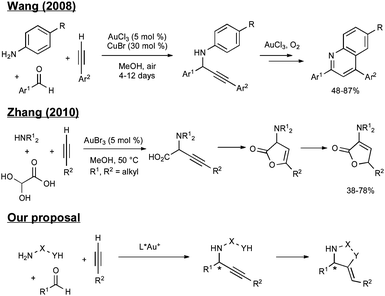 | ||
| Scheme 1 Current state-of-the-art and proposed three-component asymmetric reaction. | ||
Racemic three-component reaction
The Au(I)-catalyzed cyclization of Boc-protected propargylamines to afford five-membered carbamates with the concurrent loss of isobutylene is a known process.11 Therefore, this represented a promising arena for initial studies toward the development of a tandem process if the Boc-protected propargylamine 3 could be efficiently prepared in gold-catalyzed process. We envisioned two different strategies. First, and more simply, 3 could be prepared by the addition of an alkyne to an N-Boc imine. Alternatively, it would be formed by the addition of an alkyne to a N-aryl or -alkyl imine with subsequent acylation using standard reagents for the introduction of the Boc group (Scheme 2). Unfortunately neither strategy was ultimately successful. The Au(I)-catalyzed alkynylation of N-Boc imine 1 failed under a variety of acidic, basic, and neutral conditions. Presumably, the inability to conduct this coupling in neutral or basic conditions is due to the low reactivity of Au(I)-acetylides and unprotonated N-Boc imines (lithium and magnesium acetylides are typically used to effect this transformation). Further, when an exogenous base is not present, it is unlikely the N-Boc imine 1 is basic enough to generate the corresponding iminium and Au(I)-acetylide. The second strategy was also unsuccessful as propargylamine 5 generated from N-benzylideneaniline was remarkably resistant to Boc-protection under conditions compatible with Au(I)-catalyzed alkynylation (chlorinated or aromatic solvents, near room temperature). In direct analogy, the use of carbon dioxide in the presence of Ph3PAuCl/AgNTf2 was investigated with the hypothesis that if an equilibrium could be attained to generate trace carbamic acid 6, the reaction could be driven to completion by a subsequent and irreversible Au(I)-catalyzed cyclization. These attempts were also unsuccessful; however, this result prompted the search for a more active, yet functionally similar acylating agent.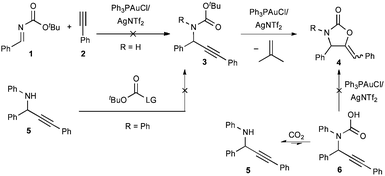 | ||
| Scheme 2 Attempts to access N-Boc propargylamines. | ||
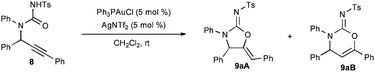
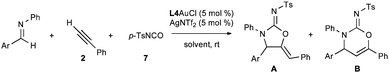
The failure of N-aryl and -alkyl isocyanates or carbodiimides to react with 5 in CH2Cl2 at room temperature illustrates the low nucleophilicity of hindered secondary anilines (Scheme 3). Fortunately, the use of the highly electrophilic p-toluenesulfonyl isocyanate (7) rapidly formed the desired urea. The urea 8 could be readily cyclized in three hours using 5 mol % Ph3PAuCl/AgNTf2 to afford a quantitative yield of the corresponding cyclic five- and six-membered carbamimidates as an 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regiomeric mixture. The connectivity and stereochemistry was ascertained through COSY and NOESY 1H-NMR experiments, the absence of a carbonyl stretching absorption in the IR spectrum, and by analogy (for the major regioisomer) to the solved crystal structure of 9pA.12
:1 regiomeric mixture. The connectivity and stereochemistry was ascertained through COSY and NOESY 1H-NMR experiments, the absence of a carbonyl stretching absorption in the IR spectrum, and by analogy (for the major regioisomer) to the solved crystal structure of 9pA.12
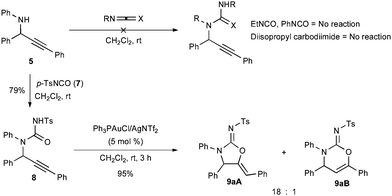 | ||
| Scheme 3 Acylation and cyclization of amine 5. | ||
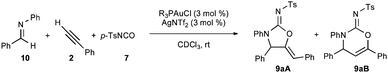
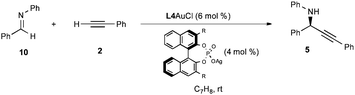
After demonstrating that the three necessary transformations (alkynylation, acylation, and cyclization) were all competent under identical reaction conditions, we sought to combine the steps to allow for a one-pot multicomponent coupling reaction between phenylacetylene (2), imine 10, and isocyanate 7 using Ph3PAuCl/AgNTf2. Gratifyingly, a 89% yield of a 15:1 mixture of 9aA:9aB was obtained after 24 h.
 | (1) |
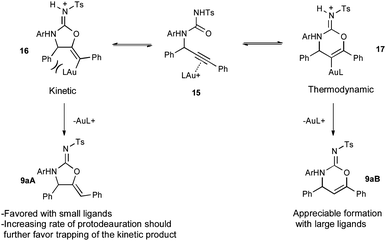
Fig. 1 shows the hypothesized catalytic cycle. Initial coordination of 2 to gold produces the alkyne π-complex 11 with acidification of the acetylenic hydrogen atom. Deprotonation by imine 10 produces the electrophilic iminium 12 with concurrent production of the Au(I)-acetylide 13. An addition reaction produces propargylamine 5 and regenerates the gold cation. Amine 5 is trapped with p-TsNCO to generate the acyclic urea 8, the alkyne of which coordinates to gold to form the second alkyne π-complex 15. 5-Exo-digcyclization by nucleophilic attack of the urea oxygen forms the vinyl gold carbamimidinium ion 16 (the minor 6-endo-dig product is not shown), which undergoes proton transfer to release the product 9aA and regenerate the Au(I) catalyst.
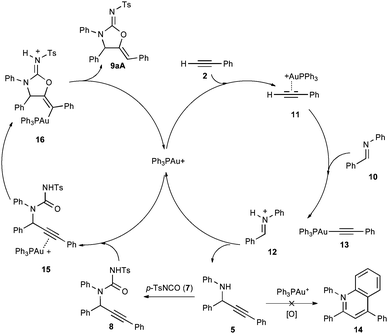 | ||
| Fig. 1 Proposed catalytic cycle. | ||
Several points about the one-pot reaction are worthy of mention. First for the addition of phenylacetylene to imine 10, the reaction time is reduced and the yield is substantially improved in the presence of isocyanate 7. When present, amine 5 can compete with the imine for protons in solution, which reduces the concentration of the reactive iminium. It can also compete with phenylacetylene as a ligand for cationic Au(I), which reduces the activity of the gold catalyst. The facile reaction between 5 and 7 to form acyclic urea 8 prevents the build up of amine 5 and avoids these detrimental interactions. Additionally, the chemical yield is improved because quinoline 14 and related hydroarylation byproducts that usually form during the alkynylation are eliminated.7h The electron rich N-arene of 5 is responsible for the high relative rate of hydroarylation; this arene is deactivated when 5 is converted to urea 8.
Conversely, the rate of cyclization is considerably slower during the multicomponent reaction. Imine 10 is fully consumed over three hours, but the reaction requires a further 45 h for complete cyclization; the cyclization was found to require only three hours when conducted in isolation. A series of control experiments was conducted in order to examine the effect of excess phenylacetylene, p-TsNCO, and p-TsNH2 during the cyclization from 8 to 9a (Table 1). Both phenylacetylene and p-TsNCO reduced the rate of cyclization by about half, while p-TsNH2 had a slightly accelerating effect. The negative effect of excess phenylacetylene is rationalized by its ability to reversibly bind to the Au(I) center and competing with the more hindered alkyne of 8. The rate retardation caused by the isocyanate 7 was unexpected and somewhat puzzling; however, further experiments (vide infra) have shown trace water present exerts an accelerating effect, potentially by acting as a proton shuttle.13 It is hypothesized that water scavenging by isocyanate 7 is to blame for the rate decrease as opposed to a direct catalyst-isocyanate interaction. Additionally, the absence of either AgNTf2 or Ph3PAuCl in the catalyst mixture was not capable of catalyzing the cyclization under the reaction conditions.
| Entry | Additive | % yield (1 h)b | % yield (3 h)b | regio (A:B)c |
|---|---|---|---|---|
| a Reactions performed on 0.14 mmol scale 8 (0.2 M) with 100 mol % of the additive in a sealed vial. b Determined with the use of an internal standard (PhNO2) by 1H NMR. c Determined by 1H NMR. d Complete decomposition observed after 20 h. | ||||
| 1 | Phenylacetylene (2) | 32 | 70 | 18:1 |
| 2 | p-TsNCO (7) | 37 | 81 | 15:1 |
| 3 | p-TsNH2 | 79 | 99 | 17:1 |
| 4 | No additive | 74 | 96 | 17:1 |
| 5 | No AgNTf2 | 0 | 0 | NA |
| 6 | No Ph3PAuCl | 0 | 0d | NA |
A brief ligand screen was conducted for the tandem three-component reaction between (E)-benzylideneaniline (10), phenylacetylene (2), and p-toluenesulfonylisocyanate (7) (Table 2). Triarylphosphines and a simple, unhindered phosphoramidite were clearly the best ligands for this transformation, providing high yields and regioselectivity for the 5- versus 6-membered cyclic urea (9aA and 9aB). Trialkylphosphines were both less active and selective, with the bulky ligand, P(tBu)3, barely providing any product after 72 h. The digold complexes of bis-phosphine ligands were also not generally able to catalyze the hydroamination step. Xantphos provided some product after an extended reaction time, but in very poor 2:1 mixture of regioisomers. All of these the catalysts were competent for the alkynylation reaction, but further increasing the steric bulk of the ligands results in a catalyst that cannot even perform the alkynylation (i.e. bulky phosphoramidites and bis-phosphines, vide infra). In general, sterically congested ligands result in a slower reaction and give diminished regioselectivity.
| Entry | Ligand | % yield (24 h)c | % yield (72 h)c | Regio (9aA:9aB)d |
|---|---|---|---|---|
| a Reactions performed on 0.55 mmol scale 10 (0.2 M) with 2 (1.2 equiv) and 7 (1.2 equiv) in a sealed vial. b 1.5 mol % of the digold catalyst used. c Determined with the use of an internal standard (mesitylene) by 1H NMR. d Determined by 1H NMR. | ||||
| 1 | PPh3 | 26 | 89 | 16:1 |
| 2 | P(p-CF3-Ph)3 | 43 | 82 | 14:1 |
| 3 | P(p-MeO-Ph)3 | 21 | 72 | 18:1 |
| 4 | P(p-Me-Ph)3 | 20 | 67 | 14:1 |
| 5 | PPh2(o-biphenyl) | 10 | ND | 16:1 |
| 6 | PCy3 | 5 | 34 | 6:1 |
| 7 | P(t-Bu)3 | 0 | 4 | NA |
| 8b | dppm | 0 | 2 | NA |
| 9 | BINAP | 0 | ND | NA |
| 10 | Xantphos | 0 | 25 | 2:1 |
| 11 |
|
65 | 92 | >20:1 |
A possible explanation for the trends observed during the ligand screen is shown in Scheme 1 under the assumption that initial cyclizations to form the vinyl gold species 16 and 17 are reversible (Fig. 2). Au-catalyzed 5-exo-digcyclization of 15 is kinetically preferred, and trapping of this vinylgold intermediate (16) by protodeauration is irreversible as the purified regioisomers are stable upon being resubjected to Ph3AuNTf2 in CH2Cl2. When small ligands are used, the rate of protodeauration to form 9aA is faster than the rate of reversion to the acyclic urea-gold complex 15 and high regioselectivities are obtained. Conversely, the ring-opening is accelerated by large ligands because of an increase in A(1,3)-strain within 16. This results in increased formation of the thermodynamically preferred 6-membered vinylgold intermediate (17) and greatly reduced the regioselectivity. It is hypothesized that the rate of protodeauration is decreased when larger ligands are used which also contributes to the erosion of regioselectivity.
 | ||
| Fig. 2 Manifestations of a reversible oxy-auration. | ||
Using this model, we surmised the regioselectivity could be enhanced if additives could be found that facilitate protodeauration: protic cosolvents and/or acids. This hypothesis is supported by experiments using N-benzylidene(2,6-difluoroaniline) (18) and the corresponding acyclic urea 19. When the multicomponent reaction is conducted with 18 using Ph3PAuNTf2, a particularly poor regioselectivity (2.4:1 of 9bA:9bB) is obtained. As mentioned previously, the presence of p-TsNCO in the medium scavenges any water present. However, conducting only the cyclization in reagent grade CH2Cl2, without the exclusion of residual water, results in an increased 6:1 ratio of 9bA:9bB. Surprisingly, the further addition of weakly acidic additives reduces the regioselectivity. Strong acids, such as methanesulfonic acid (MsOH), give the best regioselectivities, but it is unlikely that gold is involved in the cyclization when these acids are present as a control reaction shows that MsOH catalyzes the cyclization with identical selectivity in the absence of Au(I). Unlike the sulfonic acids, the weakly acidic additives shown in Table 3 are not catalytically active. Although the acid-catalyzed process results in a higher selectivity for this substrate, in all other cases examined the Ph3PAuNTf2-catalyzed cyclization imparts greater selectivity for the five-membered carbamimidate (17:1 vs. 5:1 for 9aA:9aB; 2.4:1 vs. 1:1.2 for 9qA:9qB; 1.6:1 vs. 1:1 for 9tA:9tB).
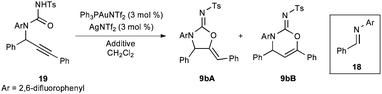
| Entry | Additive | Mol (%) | Regio (9bA:9bB)c |
|---|---|---|---|
| a Reactions performed on 0.02 mmol scale 19 (0.2 M) in a sealed vial. b Reaction performed on 0.28 mmol scale 18 (0.2 M) with 2 (1.2 equiv) and 7 (1.2 equiv) in a sealed vial. c Determined by 1H NMR. | |||
| 1b | None (from imine 18) | NA | 2.4:1 |
| 2 | None | NA | 6.0:1 |
| 3 | PhCO2H | 80 | 3.3:1 |
| 4 | 3,5-(CF3)2C6H3OH | 40 | 4:1 |
| 5 | CF3CH2OH | 40 | 4:1 |
| 6 | C6F5OH | 40 | 4.5:1 |
| 7 | PhB(OH)2 | 40 | 4.5:1 |
| 8 | MsOH | 40 | 7.5:1 |
| 9 | p-TsOH-H2O | 40 | 9:1 |
| 10 | MsOH (no Au) | 40 | 8:1 |
Standard reaction variables were briefly evaluated. Chlorinated and aromatic solvents are suitable, with chlorinated solvents providing slightly faster rates, higher yields, and improved selectivities. The effect of temperature on the regioselectivity has not been extensively studied, but, in general, modest increases in temperature up to 50 °C typically have a negligible effect.
Taking into account the aforementioned data, standard reaction conditions were designed to evaluate the scope of the multicomponent reaction. The Ph3PAuCl/AgNTf2 catalyst system was employed, because it engendered a high level of reactivity, selectivity, and catalyst stability. While the catalyst prepared from the electron-poor phosphine P(p-CF3-Ph)3 exhibited higher activity, the phosphine is nearly 1000 times more expensive per mol than PPh3. The catalyst prepared from the rac-BINOL/NMe2 phosphoramidite was also highly active, but it was found to be capricious in nature and reactions frequently stalled prior to complete conversion. Chloroform was chosen as a solvent over methylene chloride only because its use allows a larger range of temperatures to be achieved without resorting to superheating; the results were otherwise essentially identical.
The scope of aryl-aryl imines is broad, providing high yields and regioselectivities for a variety of electronically and sterically diverse substrates (Table 4). Exceptions to the latter points include the imines prepared from benzaldehyde and 3,5-bis(trifluoromethyl)aniline and 2,6-difluoroaniline (to provide products 9o and 9b). The low regioselectivity of the former is consistent with the model advanced in Fig. 2; the powerful inductive effect of the electron-poor N-aryl group enables a facile retroauration of vinyl gold 16 and results in a substantial quantity of the thermodynamically preferred six-membered carbamimidate. It has been established in the latter case that the regioselectivity can be increased to 8:1 by using an acid additive (MsOH). Both the benzaldehyde and aniline-derived arenes tolerate substitution at ortho, meta, and para positions; however, several substitution patterns were not amenable to the multicomponent reaction (Fig. 3). Imines 20 and 21, bearing large ortho substituents, were unreactive toward the Au(I)-acetylide and undergo slow decomposition. Conversely, the small ortho-cyano substituent in 22 impedes the cyclization but not the alkynylation. The placement of meta-alkoxy substituents (23, 24, and 25) on the N-aryl ring increases the nucleophilicity of the arene sufficiently that a Friedel–Crafts reaction with the iminium is faster than alkynylation. Thus, these imines predominantly form oligomers.
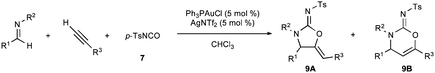
| Entry | R1 | R2 | R3 | Product | T/°C | Time (h) | % yield XAb | regio (A:B)d |
|---|---|---|---|---|---|---|---|---|
| a Reactions performed on 0.28 mmol or 0.50 mmol scale imine (0.2 M) with alkyne (1.2 equiv) and 7 (1.2 equiv) in a sealed vial. b Isolated yields of major isomer unless otherwise specified. c Isolated as a mixture of regioisomers. d Determined by 1H NMR of the crude reaction mixture. | ||||||||
| 1 | Ph | Ph | Ph | 9a | 35 | 20 | 84 | 17:1 |
| 2 | p-tolyl | Ph | Ph | 9c | 35 | 20 | 80 | >20:1 |
| 3 | m-tolyl | Ph | Ph | 9d | 35 | 20 | 83 | >20:1 |
| 4 | o-tolyl | Ph | Ph | 9e | 35 | 20 | 79 | 17:1 |
| 5 | 2-napthyl | Ph | Ph | 9f | 35 | 20 | 78 | 19:1 |
| 6 |

|
Ph | Ph | 9g | 35 | 20 | 85c | 13:1 |
| 7 |

|
Ph | Ph | 9h | 35 | 20 | 81 | 14:1 |
| 8 |
|
Ph | Ph | 9i | rt | 72 | 68c | 13:1 |
| 9 |

|
Ph | Ph | 9j | 35 | 20 | 80 | 14:1 |
| 10 | Ph |

|
Ph | 9k | 35 | 20 | 80 | 19:1 |
| 11 | Ph |
|
Ph | 9l | 35 | 48 | 76 | 16:1 |
| 12 | Ph |
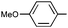
|
Ph | 9m | 35 | 20 | 73 | >20:1 |
| 13 | Ph |
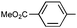
|
Ph | 9n | 35 | 48 | 75 | 18:1 |
| 14 | Ph |
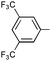
|
Ph | 9o | 35 | 48 | 65 | 5:1 |
| 15 | Ph |
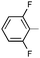
|
Ph | 9b | rt | 6 d | 55 | 2.4:1 |
| 16 | Ph |
|
Ph | 9p | rt | 48 | 75 | 17:1 |
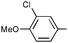
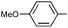
| 17 | Ph | Ph |
|
9q | rt | 48 | 45 | 2.4:1 |
| 18 | Ph | Ph |
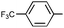
|
9r | 50 | 48 | 90 | >20:1 |
| 19 | Ph | Ph | Bn | 9s | 50 | 48 | 72 | 15:1 |
| 20 | Ph | Ph | n-Bu | 9t | 50 | 48 | 47 | 1.6:1 |
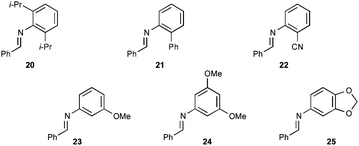 | ||
| Fig. 3 Unsuitable imine substrates. | ||
Three alkyne types were evaluated: aryl, alkyl, and silyl. An electron-rich aromatic alkyne, p-methoxyphenylacetylene, reacted readily at room temperature to give the cyclic urea 9q in modest yield and regioselectivity while an electron-poor aryl alkyne required 50 °C, but provided a nearly quantitative yield of a single regioisomer 9rA. The aliphatic alkynes 1-hexyne and 3-phenylpropyne both required an elevated temperature for the reaction to occur at a reasonable rate. The differences between the two are surprising, as the reaction using 3-phenylpropyne is higher yielding and drastically more regioselective. TMS-acetylene did not react with the imine, even under forcing conditions.
Enantioselective alkynylation
In order to develop an enantioselective variant of the multicomponent reaction, we were confronted with a significant limitation with regards to ligand selection. The vast majority of Au(I)-catalyzed asymmetric reactions developed have utilized enantioenriched axially chiral bisphosphine ligands (i.e.BINAP, SEGPHOS, and BIPHEP derivatives),14 although reports using chiral phosphoramidites/phosphites,15 and N-heterocyclic carbene16 ligands have also been published. We were forced to exclude potential candidates from these classes because earlier ligand screening revealed that even the relatively unhindered parent BINAP ligand was unable to catalyze 5-exo-digcyclization. We hypothesized chiral 1-diarylphosphino-2-amino cyclohexane derivatives, particularly ureas, would be sufficiently small to allow the cyclization to occur and provide a scenario to merge hydrogen-bond and gold catalysis. Because the reactive electrophile is the iminium formed by deprotonation of the Au(I) acetylene π-complex by the imine, traditional H-bond donor catalysis would likely not be operative. Instead, we reasoned a closed, ordered transition state might result from the urea functioning instead as an H-bond acceptor. The bound iminium is necessarily less electrophilic than the free iminium; therefore, this manifold requires this reduced electrophilicity is compensated by an entropic advantage obtained from bringing the two reactive species (iminium and Au(I) acetylide) together.We began our investigations using trans-1-diarylphosphino-2-amino cycloalkanes. A simple derivative, trans-1-diphenylphosphino-2-aminocyclohexane (26), has been prepared in optically pure form by Jacobsen using a tartaric acid resolution of the racemate.17 From 26, a variety of gold catalysts were easily synthesized by derivatization of the amine and ligand exchange with Me2S·AuCl (eqn (2)).
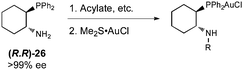 | (2) |
Initially, thiourea-containing ligands were prepared, but the corresponding digold complexes provided propargyl amine 5 as a racemic mixture. However, the use of the monogold complex of L1 containing a sulfonylurea led to a promising 65% ee, and a wide range of ligands bearing various N-substituents was prepared and screened in the model reaction (Fig. 4). It was initially apparent that although ureas, in particular sulfonylureas (L1–L4), give the highest enantioselectivities, other functionalities such as carbamates (L5–L7) and amides (L17, L18) can provide moderate to good levels of stereocontrol. Sulfonamides L20 and L21 and the phthalate L19 were particularly poor catalysts. The unsubstituted amine was not a competent catalyst, but this is likely due to poor solubility in the reaction medium as other primary amines bearing greasy P-aryl substituents did provide the propargyl amine with some selectivity (vide infra). A significant effort was made to prepare the chiral sulfinylurea analogues of L1 and L4 with the hope of observing an additive effect between the sulfinyl stereocenter and the ligand backbone to achieve greater selectivities.18 Unfortunately, these ligands were not able to be synthesized using a variety of methods. It is not completely clear why this is the case, but 31P-NMR of the post-reaction mixture showed several phosphine oxides to be present. It is possible internal redox chemistry occurs between the sulfinylurea and the phosphine. In this light, it was hoped an equivalent carbon stereocenter, derived from an amino acid, would mimic the sought after chiral sulfinylureas. Ligands derived from tert-leucine, L12 and L13, represented a matched and mismatched case, respectively, but the matched case provided amine 5 in only 45% ee.
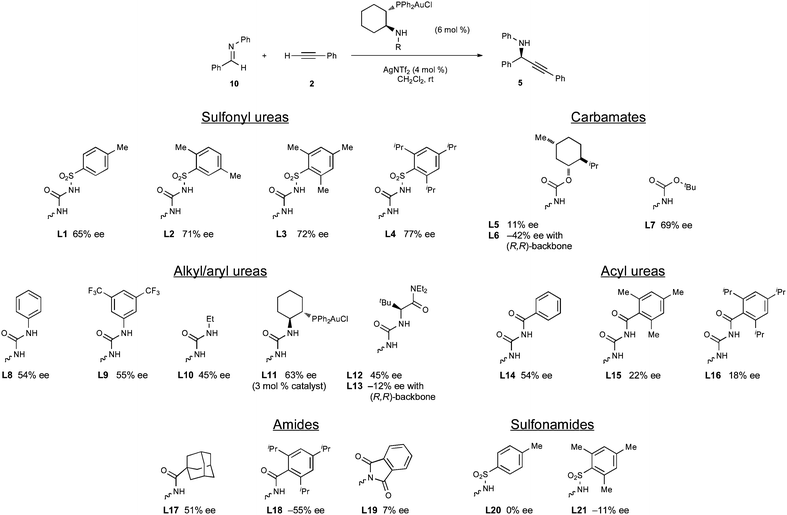 | ||
| Fig. 4 Effect of N-substitution. | ||
With some data in hand regarding the effect of N-substitution, we next sought to evaluate ligands containing alternative P-aryl groups. The ligands were synthesized through the ring-opening reaction of enantiomerically-enriched cyclic sulfamidates with metal phosphides developed recently by Guo and coworkers.19,20 The use of this method did increase the overall length of the ligand syntheses, but avoided the tedious resolution of individual ligands.
Unfortunately, the fourteen p-toluenesulfonylurea ligands prepared all gave inferior results relative to L1 (Table 5). Some general trends were established: 1. the cyclohexane skeleton provides better selectivity than the cyclopentane ligands which is in turn generally better than the indane ligands; 2. the enantioselectivity decreases for a given class upon increasing the steric bulk of the P-aryl substituents; 3. electron-rich P-aryl groups provide higher enantioselectivities than electron-poor P-aryl groups when the sterics are roughly equal.
| Ar | % eeb using
|
% eeb using
|
% eeb using
|
|---|---|---|---|
| a Reactions performed on 0.03 mmol scale 10 (0.2 M) with 2 (2.0 equiv) for 2–6 h in a sealed vial. R = C(O)NHTs. b Determined by chiral HPLC. | |||
| Ph | −65 (L1) | −48 (L22) | −36 (L23) |
| 4-CF3-C6H4 | –59 (L24) | NA | NA |
| 2-Me-C6H4 | −47 (L25) | −37 (L26) | −23 (L27) |
| 3,5-Me2-C6H3 | −53 (L28) | −38 (L29) | −40 (L30) |
| 3,5-(CF3)2-C6H3 | −37 (L31) | −2 (L32) | −17 (L33) |
| 3,5-(tBu)2-4-MeO-C6H2 | −35 (L34) | −11 (L35) | NA |

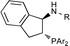
There is evidence that sulfonyl ureas are not the optimal N-analog for all of the scaffolds examined. For example, within the DTBM series, the selectivity steadily increased as the size of the acyl group decreased (Fig. 5). The use of the simple primary amine L38 was the most selective ligand, but still provided amine 5 in only −57% ee. There may be a balance that must be achieved between the sterics of the P- and N-substituents; increasing the size of one requires a corresponding decrease in the size of the other to obtain the best selectivity. Regardless, further optimization studies were carried out using the 2,4,6-triisopropylsulfonyl (trisyl) urea catalyst L4, because it provided the best selectivity (77% ee) and is easily synthesized from the parent scaffold.
 | ||
| Fig. 5 Effect of N-substitution within the DTBM ligand series. | ||
To begin, the common reaction parameters (solvent, counterion, temperature, catalyst loading and concentration) were evaluated using L4. It was found that toluene was the optimal solvent and increased the selectivity to 85% ee. Other chlorinated and aromatic solvents used were generally competent but not quite as selective (73–82% ee). The counterion of choice was bistriflimide (Tf2N−), though SbF6−, PF6−, TfO−, BF4−, and TsO− were nearly as selective. No reaction was observed with more coordinating counterions with the exception of diarylphosphates (vide supra). Changing the temperature or the catalyst loading had no effect, whereas the selectivity was decreased upon dilution of the reaction. The highest selectivity was found when the starting concentration of the imine was at or above 0.2 M.
In the light of recent reports from our lab detailing highly enantioselective Au(I) catalysis using chiral BINOL-derived phosphate counterions, this strategy was evaluated for the asymmetric alkynylation.21 No alkynylation was observed with using Ph3PAu(I)-chiral phosphate complexes (prepared in situ from Ph3PAuCl and a silver phosphate). Surprisingly, the alkynylation did proceed, albeit slowly, when using (R,R)-L4AuCl as the precatalyst although the enantioselectivity was not increased above that obtained using (R,R)-L4AuNTf2 (85% ee in toluene) (Table 6). The reaction failed when L10 was used, containing a more electron-rich urea. Finally, because the cyclization reaction was not catalyzed by these Au(I) phosphate complexes, we did not evaluate them further as potential catalysts for the tandem reaction.
| Entry | Ligand | R = | % eeb |
|---|---|---|---|
| a Reactions performed on 0.03 mmol scale 10 (0.2 M) with 2 (2.0 equiv) for 2–6 h in a sealed vial. b Determined by chiral HPLC. | |||
| 1 | (R,R)-L4 | adamantyl | –83 |
| 2 | (S,S)-L4 | adamantyl | 81 |
| 3 | (R,R)-L4 | 2,4,6-iPr3-C6H3 | 33 |
| 4 | (S,S)-L4 | 2,4,6-iPr3-C6H3 | 77 |
Using the conditions optimized for enantioselectivity (L4AuCl/AgNTf2, 0.2 M imine, toluene, rt), the three-component reaction was evaluated using (E)-benzylideneaniline, phenylacetylene, and p-toluenesulfonylisocyanate. It was immediately apparent that the use of the large ligand L4 was detrimental to the cyclization with regards to the reaction rate, reaction efficiency, and regioselectivity (entry 1, Table 7). Switching to methylene chloride resulted in a 75% yield after 48 h but did not significantly improve the regioselectivity (entry 2). The use of (E)-4-chlorobenzylideneaniline, of which the acyclic urea intermediate is less reactive during the cyclization, illustrates the limitations of this system (entries 3 and 4). Even after a prolonged reaction time, full conversion of the acyclic urea is not attained.
| Entry | Ar | Solvent | Time (h) | % yieldb | regio (A:B)c | % eed |
|---|---|---|---|---|---|---|
| a Reactions performed on 0.03 mmol scale imine (0.2 M) with 2 (1.2 equiv) and 7 (1.2 equiv) in a sealed vial. b Determined with the use of an internal standard (mesitylene) by 1H NMR. The value in parentheses is the yield of the corresponding uncyclized urea. c Determined by 1H NMR. d Determined by chiral HPLC. | ||||||
| 1 | Ph | C7H8 | 21 | 27 (37) | 4.5:1 |
85 |
| 2 | Ph | CH2Cl2 | 48 | 75 (0) | 5:1 |
81 |
| 3 | 4-Cl-C6H4 | CH2Cl2 | 24 | 30 (51) | 3:1 |
83 |
| 3 | 4-Cl-C6H4 | CH2Cl2 | 168 | 52 (14) | 3:1 |
82 |
In order to address the sluggish cyclization, information obtained during studies of the racemic reaction was reevaluated. In particular, some data collected suggested the presence of water may increase the regioselectivity and rate of cyclization, although it was not necessary to exploit that finding to achieve an effective racemic reaction. Since the same problems with increased severity resurfaced in the asymmetric variant, the addition of water post-alkynylation was evaluated. Ultimately, we found the addition of 0.5 equivalents of water after the complete consumption of imine greatly accelerated the cyclization allowing a reduction in the catalyst loading from 10 to 5 mol % and roughly doubling the regioselectivity (Table 8). The enantioselectivity was not significantly affected for most substrates. This modification was not amenable to substrates in which the alkynylation was accompanied by significant quantities of byproducts (entries 9 and 16). In these cases, the addition of water released various basic compounds (amines) that completely shut down the cyclization reaction. Some substrates were converted with greater enantioselectivity without the addition of water (entries 5, 10, and 12), while in other cases where the rate of cyclization was comparable with the rate of alkynylation, cyclization was essentially complete by the time the imine was fully consumed (entries 17 and 18). Although toluene provided the best enantioselectivity during optimization studies, the addition of water post-alkynylation to reactions conducted in toluene did not lead to efficient cyclizations. This is potentially because the alkynylations did not occur as cleanly. In general, good to excellent % ee's were obtained from a variety of aryl-aryl imines. Imines derived from ortho-substituented benzaldehydes (entries 5,7, and 8) or 3,5-disubstituted anilines (entries 9 and 13) were converted with the highest enantioselectivities.
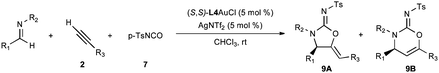
| Entry | R1 | R2 | R3 | Product | Method | Time (h)b | % yield 9Ac | % ee 9Ad | Regio (9A:9B)e |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed on 0.14 mmol or 0.061 mmol scale imine (0.2 or 0.4 M) with alkyne (1.2 equiv) and 7 (1.2 equiv) in a sealed vial. Method A = No modification to the reaction conditions. Method B = After complete conversion of the imine by TLC analysis, H2O (0.5 equiv) in CHCl3 was added and let stand for the time indicated. b Time 4 + 36 refers to 4 h prior to the addition of H2O and 36 h after the addition of H2O. c Isolated yields of major isomer unless otherwise specified. d Determined by chiral HPLC. e Determined by 1H NMR of the crude reaction mixture. f (R,R)-L4AuCl (10 mol %) and AgNTf2 (10 mol %) were used. g Reaction conducted in CH2Cl2. h Reaction conducted in C7H8. i Reaction was conducted with alkyne (2.0 equiv) and 7 (2.0 equiv). j Isolated as a mixture of regioisomers. k Yield determined with the use of an internal standard (PhNO2) by 1H NMR. | |||||||||
| 1 | Ph | Ph | Ph | (R)-9a | B | 4 + 36 | 76 | 79 | 8:1 |
| 2 | p-tolyl | Ph | Ph | (R)-9c | B | 4 + 36 | 76 | 80 | 11:1 |
| 3 | m-tolyl | Ph | Ph | (R)-9d | B | 4 + 36 | 75 | 68 | 8:1 |
| 4 | 2-napthyl | Ph | Ph | (R)-9f | B | 4 + 36 | 68 | 62 | 9:1 |
| 5 |

|
Ph | Ph | (R)-9g | A | 6 d | 60j | 82 | 7:1 |
| 6 |
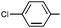
|
Ph | Ph | (R)-9h | B | 4 + 36 | 63 | 79 | 6:1 |
| 7 |
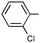
|
Ph | Ph | (R)-9i | B | 4 + 36 | 61j | 91 | 7:1 |
| 8 |
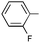
|
Ph | Ph | (R)-9j | B | 4 + 36 | 55 | 86 | 8:1 |
| 9 | Ph |
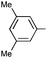
|
Ph | (S)-9k | Af,g | 96 | 38 | 91 | 3:1 |
| 10 | Ph |
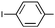
|
Ph | (S)-9l | Af,g | 96 | 43 | 84 | 3:1 |
| 11 | Ph |
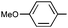
|
Ph | (R)-9m | B | 4 + 36 | 69 | 72 | 8:1 |
| 12 | Ph |
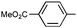
|
Ph | (S)-9n | Af,g | 96 | 45 | 78 | 4:1 |
| 13 | Ph |
|
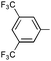
Ph | (R)-9o | B | 4 + 36 | 48 | 95 | 5:1 |
| 14 | Ph |
|
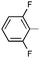
Ph | (S)-9b | Af | 6 d | 48 | 41 | 1.7:1 |
| 15 | Ph |
|
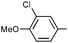
Ph | (R)-9p | B | 4 + 36 | 70 | 77 | 6:1 |
| 16 | Ph | Ph |
|
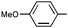
(S)-9q | Af,i | 5 d | 25k | 74 | 1:1.8 |
| 17 | Ph | Ph |
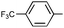
|
(R)-9r | Ah | 6 d | 85 | 72 | >20:1 |
| 18 | Ph | Ph | Bn | (R)-9s | Ah | 6 d | 66 | 76 | 6:1 |
It is unclear what interactions are responsible for the high facial selectivities imparted by the trans-1-diarylphosphino-2-aminocycloalkane ligands, in particular L4. It is possible that the ligand-Au(I) acetylide complex coordinates to the incoming iminium ion by hydrogen bonding through the carbonyl functionality of the amine derivative. Such an interaction is consistent with the findings that ligands containing ureas, amides, or carbamates provide all provide at least moderate selectivities, while those containing considerably less basic moieties (sulfonamides, phthalate) provide nearly racemic amines. Alternatively, through serendipity the complexes may simply possess an appropriate topology that differentiates between the two enantiotopic faces of the iminium ion. Regardless of the origin of selectivity, this is the first report of a chiral ligand-Au(I) complex that catalyzes the activation and transfer of an alkyne (presumably through the Au(I)-acetylide) to an electrophile.
Conclusions
We have identified a system in which a single gold species can catalyze both the alkynylation of aryl-aryl imines and the subsequent 5-exo-digcyclization of the corresponding acylic urea to provide cyclic five-membered carbamimidates. Although similar gold-catalyzed tandem reactions have been disclosed, this is the first to preserve the stereocenter set in the alkynylation through the sequence. This feature was exploited through the development of an asymmetric three-component reaction. Because traditional biaryl bisphosphine ligands were not competent in the cyclization step, a fundamentally new class of ligands for asymmetric Au(I) catalysis was necessary.22 To fulfil this need, we have developed and utilized trans-1-diarylphosphino-2-aminocycloalkanes, which provide the product cyclic five-membered carbamimidates in moderate to high regio- and enantioselectivities. Additional research may shed light upon the key interactions responsible for the high facial selectivities obtained as well as expand the use of these ligands to other asymmetric Au(I)-catalyzed reactions.Acknowledgements
We gratefully acknowledge NIHGMS (RO1 GM073932-05) for financial support. X-ray crystallography was performed by Dr Antonio DiPasquale. We thank Dr Nathan Shapiro and Dan Gray for conducting preliminary experiments regarding asymmetric alkylylations, and Johnson-Matthey for a generous gift of AuCl3.Notes and references
- C. Mannich and Kröschl, Arch. Pharm., 1912, 250, 647 CrossRef.
- I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1, 8–21 CrossRef.
- P. Biginelli, Ber. Dtsch. Chem. Ges., 1891, 24, 1317 CrossRef; P. Biginelli, Ber. Dtsch. Chem. Ges., 1891, 24, 2962 CrossRef; P. Biginelli, Ber. Dtsch. Chem. Ges., 1893, 26, 447 CrossRef.
- M. Passerini, Gazz. Chem. Ital., 1921, 51, 126–129 Search PubMed; M. Passerini, Gazz. Chem. Ital., 1921, 51, 181–183 Search PubMed.
- For reviews of multicomponent reactions, see: J. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed; A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 Search PubMed.
- For recent developments in gold catalysis, see: (a) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358–1367 RSC; (b) A. Furstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef CAS; (d) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS; (e) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS; (f) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS; (g) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC; (h) H. C. Shen, Tetrahedron, 2008, 64, 3885–3903 CrossRef CAS; (i) R. Skouta and C.-J. Li, Tetrahedron, 2008, 64, 4917–4938 CrossRef CAS; (j) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS.
- (a) C. J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS; (b) V. K. Y. Lo, K. K. Y. Kung, M. K. Wong and C. M. Che, J. Organomet. Chem., 2009, 694, 583–591 CrossRef CAS; (c) V. K. Y. Lo, Y. G. Liu, M. K. Wong and C. M. Che, Org. Lett., 2006, 8, 1529–1532 CrossRef CAS; (d) C. M. Wei and C. J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS; (e) B. Yan and Y. H. Liu, Org. Lett., 2007, 9, 4323–4326 CrossRef CAS; (f) Q. Zhang, M. Chang, X. Y. Hu, B. G. Li and J. X. Ji, J. Am. Chem. Soc., 2010, 132, 7256–7257 CrossRef CAS; (g) X. Zhang and A. Corma, Angew. Chem., Int. Ed., 2008, 47, 4358–4361 CrossRef CAS; (h) F. P. Xiao, Y. L. Chen, Y. Liu and J. B. Wang, Tetrahedron, 2008, 64, 2755–2761 CrossRef CAS.
- G. Q. Tian and M. Shi, Org. Lett., 2007, 9, 4917–4920 CrossRef CAS; C. Wang, Z. Y. Han, H. W. Luo and L. Z. Gong, Org. Lett., 2010, 12, 2266–2269 CrossRef CAS.
- A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS.
- For recent reviews of asymmetric gold-catalyzed reactions, see: A. Pradal, P. Y. Toullec and V. Michelet, Synthesis, 2011 DOI:10.1055/s-0030-1258465; S. Sengupta and X. D. Shi, ChemCatChem, 2010, 2, 609–619 Search PubMed; R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382–5391 Search PubMed.
- A. Buzas and F. Gagosz, Synlett, 2006, 2727–2730 CAS; A. K. Buzas, F. M. Istrate and F. Gagosz, Tetrahedron, 2009, 65, 1889–1901 CrossRef CAS; E. S. Lee, H. S. Yeom, J. H. Hwang and S. Shin, Eur. J. Org. Chem., 2007, 3503–3507 CrossRef CAS; R. Robles-Machin, J. Adrio and J. C. Carretero, J. Org. Chem., 2006, 71, 5023–5026 CrossRef CAS.
- CCDC XXXXX contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
- The data does not rule out the possibility that a chiral Brønsted acid is formed from the interaction of water and the Au(I) catalyst which is responsible for catalyzing the cyclization.
- S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276–8277 CrossRef CAS; M. Murai, J.Ä. Uenishi and M. Uemura, Org. Lett., 2010, 12, 4788–4791 CrossRef CAS; A. Martínez, P. García-García, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Angew. Chem., Int. Ed., 2010, 49, 4633–4637 CrossRef CAS; M. Martìn-Rodrìguez, C. Nojera, J. M. Sansano and F.-L. Wu, Tetrahedron: Asymmetry, 2010, 21, 1184–1186 CrossRef CAS; F. Liu, D. Y. Qian, L. Li, X. L. Zhao and J. L. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6669–6672 CrossRef CAS; R. L. Lalonde, J. Z. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 CAS; Z. Zhang, S. D. Lee and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 5372–5373 CrossRef CAS; I. D. G. Watson, S. Ritter and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2056–2057 CrossRef CAS; M. Uemura, I. D. G. Watson, M. Katsukawa and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 3464–3465 CrossRef CAS; F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS; A. S. K. Hashmi, M. Hamzic, F. Rominger and J. W. Bats, Chem.–Eur. J., 2009, 15, 13318–13322 CAS; C. M. Chao, D. Beltrami, P. Y. Toullec and V. Michelet, Chem. Commun., 2009, 6988–6990 RSC; C.-M. Chao, M. R. Vitale, P. Y. Toullec, J.-P. GenÍt and V. Michelet, Chem.–Eur. J., 2009, 15, 1319–1323 CrossRef CAS; M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9533–9537 CrossRef CAS; S. G. Sethofer, S. T. Staben, O. Y. Hung and F. D. Toste, Org. Lett., 2008, 10, 4315–4318 CrossRef CAS; Z. Zhang and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2007, 46, 283–285 CrossRef CAS; Z. Zhang, C. F. Bender and R. A. Widenhoefer, Org. Lett., 2007, 9, 2887–2889 CrossRef CAS; Z. Zhang, C. F. Bender and R. A. Widenhoefer, J. Am. Chem. Soc., 2007, 129, 14148–14149 CrossRef CAS; M. A. Tarselli, A. R. Chianese, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 6670–6673 CrossRef CAS; A. D. Melhado, M. Luparia and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12638–12639 CrossRef CAS; M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402–12403 CrossRef CAS; C. Liu and R. A. Widenhoefer, Org. Lett., 2007, 9, 1935–1938 CrossRef CAS; R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452–2453 CrossRef CAS; M. P. Muñoz, J. Adrio, J. C. Carretero and A. M. Echavarren, Organometallics, 2005, 24, 1293–1300 CrossRef CAS; M. J. G. Johansson, D. J., S. T. Staben and F.D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS; A. C. Camino González-Arellano, Marta Iglesias and Felix Sánchez, Chem. Commun., 2005, 3451–3453 RSC; Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405–6406 CrossRef CAS.
- H. Teller, S. Flügge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949–1953 CAS; A. Z. Gonzalez and F. D. Toste, Org. Lett., 2010, 12, 200–203 CrossRef; I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020–13030 CrossRef CAS.
- Y. Matsumoto, K. B. Selim, H. Nakanishi, K.-i. Yamada, Y. Yamamoto and K. Tomioka, Tetrahedron Lett., 2010, 51, 404–406 CrossRef CAS.
- Y.-Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660–5661 CrossRef CAS.
- K. L. Kimmel, M. T. Robak and J. A. Ellman, J. Am. Chem. Soc., 2009, 131, 8754–8755 CrossRef CAS.
- R. Guo, S. Lu, X. Chen, C.-W. Tsang, W. Jia, C. Sui-Seng, D. Amoroso and K. Abdur-Rashid, J. Org. Chem., 2009, 75, 937–940.
- Synthetic details are provided in the supporting information.
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS; R. L. Lalonde, J. Z. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 CAS.
- Phosphoramidites have also recently emerged as alternative ligands to biaryl bisphosphines. See references cited in 15 above.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference number 823588. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00160d |
| This journal is © The Royal Society of Chemistry 2011 |