A copper and chromium based nanoparticulate oxide as a noble-metal-free cocatalyst for photocatalytic water splitting†
Kazuhiko
Maeda
ab,
Tomoyuki
Ohno
a and
Kazunari
Domen
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp; Fax: +81-3-5841-8838; Tel: +81-3-5841-1652
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama, 332-0012, Japan
First published on 12th May 2011
Abstract
Nanoparticulate oxides consisting of copper (Cu) and chromium (Cr) were studied as noble-metal-free cocatalysts for photocatalytic water splitting. The structure of the Cu–Cr mixed oxide dispersed on a solid solution of GaN and ZnO (referred to as GaN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnO hereafter) was characterized by high-resolution transmission electron microscopy (HR-TEM), X-ray absorption fine-structure (XAFS) spectroscopy, and electrochemical measurements. The mixed-oxide nanoparticle was an effective promoter of photocatalytic overall water splitting on GaN:ZnO, and was loaded by impregnation from an aqueous solution containing Cu(NO3)2·3H2O and Cr(NO3)3·9H2O followed by calcination in air. Impregnation of GaN:ZnO with 1.5 wt% Cu and 2.0 wt% Cr followed by calcination at 623 K for 1 h provided the highest photocatalytic activity, while catalysts modified with either Cu-oxide or Cr-oxide showed little activity. The activity of this photocatalyst was shown to be strongly dependent on the generation of Cu(II)–Cr(III) mixed-oxide nanoparticles with optimal composition and coverage. The results of electrochemical measurements and photocatalytic reactions also indicated that Cu(II)–Cr(III) mixed-oxide nanoparticles on GaN:ZnO are resistant to both the photoreduction of O2 and water formation from H2 and O2, which are undesirable reverse reactions in overall water splitting.
:ZnO hereafter) was characterized by high-resolution transmission electron microscopy (HR-TEM), X-ray absorption fine-structure (XAFS) spectroscopy, and electrochemical measurements. The mixed-oxide nanoparticle was an effective promoter of photocatalytic overall water splitting on GaN:ZnO, and was loaded by impregnation from an aqueous solution containing Cu(NO3)2·3H2O and Cr(NO3)3·9H2O followed by calcination in air. Impregnation of GaN:ZnO with 1.5 wt% Cu and 2.0 wt% Cr followed by calcination at 623 K for 1 h provided the highest photocatalytic activity, while catalysts modified with either Cu-oxide or Cr-oxide showed little activity. The activity of this photocatalyst was shown to be strongly dependent on the generation of Cu(II)–Cr(III) mixed-oxide nanoparticles with optimal composition and coverage. The results of electrochemical measurements and photocatalytic reactions also indicated that Cu(II)–Cr(III) mixed-oxide nanoparticles on GaN:ZnO are resistant to both the photoreduction of O2 and water formation from H2 and O2, which are undesirable reverse reactions in overall water splitting.
Introduction
To address the depletion of fossil fuels and the relevant environmental concerns, there is an urgent need for a sustainable energy source for human society.1 Photocatalytic water splitting using semiconductor particles has long been studied as a means of producing H2 as a clean, renewable energy carrier.2 Although numerous efforts have been made in the development of photocatalytic water splitting systems, there still remain many challenges to be overcome.The reaction occurs in three steps:2b (1) the photocatalyst absorbs photon energy greater than the band-gap energy of the material and generates photoexcited electron-hole pairs in the bulk, (2) the photoexcited carriers separate and migrate to the surface without recombination, and (3) adsorbed species are reduced and oxidized by the photogenerated electrons and holes to produce H2 and O2, respectively. The first two steps strongly depend on the structural and electronic properties of the photocatalyst, while the third step is promoted by the presence of an additional catalyst (a so-called cocatalyst). It is therefore important to develop a photocatalyst and a cocatalyst in harmony.
To obtain a measurable activity, a cocatalyst must usually be loaded onto a photocatalyst.2b The loaded cocatalyst is usually in the form of nanoparticles, and is used to collect photogenerated charge carriers and host active sites for catalyticwater reduction and/or oxidation. As a result, the water splitting rate over a given photocatalyst can be improved significantly. It has been reported that NiOx,3RuO2,4 Rh2−yCryO3,5 and noble-metal/Cr2O3 (core/shell) nanoparticles6 function as effective H2-evolution cocatalysts for photocatalytic overall water splitting.
For practical application in the future, our group suggested that an area of 250000 km2, corresponding to 1% of the earth's desert area, would be required to provide one-third of the projected energy needs of human society in 2050 from solar energy, with a conversion efficiency of 10%, assuming an integrated solar energy of AM1.5G irradiation for a day with correction for sunlight angle.2f Particulate photocatalyst systems are considered to be advantageous toward such a large-scale application although a method for separating the simultaneously produced H2 and O2 remains to be developed.
It is therefore important to develop a cheap, high-performance cocatalyst as well as such a photocatalyst, because the price of a given photocatalytic material will become a key issue when the material will be employed on a large-scale. Unfortunately, these cocatalysts, except for NiOx, are only present in limited reserves on earth. In this regard, finding a new function for a commonly available material is one appealing approach. Although MoS2,7WC,8 and NiS9 have recently been reported as noble-metal-free cocatalysts for sacrificial H2 evolution with metal sulfide photocatalysts, there remains room for the development of similar inexpensive cocatalysts for non-sacrificial water splitting. Developing an earth-abundant metal-based cocatalyst that efficiently catalyzes water splitting is also a fundamental challenge in the field of artificial photosynthesis for large-scale industrial application.10 These considerations stimulated our development of an inexpensive cocatalyst that efficiently promotes water splitting.
In our previous paper, we briefly mentioned that the water splitting activity of GaN:ZnO modified with both Cu and Cr species as H2 evolution sites, was higher than that achieved by either Cu or Cr modification alone.11 However, the detailed structure and function of this Cu–Cr cocatalyst remained unclear. The present work was therefore a more detailed study of a copper–chromium noble-metal-free cocatalyst for photocatalytic water splitting using GaN:ZnO as a photocatalyst. The structural characteristics of the Cu–Cr mixed-oxide cocatalyst on the GaN:ZnO surface were investigated by XAFS in an attempt to clarify the relationship between the structure of the catalyst and the photocatalytic activity for overall water splitting. The function of Cu and Cr-based cocatalysts on GaN:ZnO is discussed on the basis of photocatalytic reaction results and electrochemical measurements.
Results and discussion
Effect of Cu and Cr loading on photocatalytic activity
Table 1 lists the rates of H2 and O2 evolution in water splitting reactions with UV irradiation (λ > 300 nm) using GaN:ZnO calcined at 623 K after impregnation of Cu(NO3)2·3H2O and Cr(NO3)3·9H2O with different amounts of Cu and Cr. In all cases, nearly stoichiometric H2 and O2 evolution (H2/O2 ≈ 2) was observed, indicating that overall water splitting did occur. At a given Cu loading, the water splitting rate increased with increasing Cr pairing, up to a certain point, beyond which the rate began to decrease. A similar trend was observed when the loading amount of Cu was changed with a fixed Cr loading. The highest activity was obtained with a catalyst modified with 1.5 wt% Cu and 2.0 wt% Cr. Note that GaN:ZnO alone produced no H2 or O2 under the present reaction conditions.11
:ZnO prepared with various loadings of Cu and Cr and a calcination temperature of 623 Ka
| Entry | Loading amount/wt% | Gas evolution rateb/μmol h−1 | ||
|---|---|---|---|---|
| Cu | Cr | H2 | O2 | |
| a Reaction conditions: catalyst, 0.3 g; distilled water, 400 mL; light source, high-pressure mercury lamp (450 W); reaction vessel, Pyrex inner-irradiation type; irradiation wavelength, λ > 300 nm. b Average rate of gas evolution in 5 h. | ||||
| 1 | 0.5 | 0.5 | 154 | 75 |
| 2 | 1.0 | 205 | 103 | |
| 3 | 1.5 | 118 | 61 | |
| 4 | 1.0 | 0.5 | 150 | 67 |
| 5 | 1.0 | 585 | 292 | |
| 6 | 1.5 | 363 | 185 | |
| 7 | 1.5 | 1.0 | 476 | 242 |
| 8 | 1.5 | 561 | 287 | |
| 9 | 2.0 | 668 | 342 | |
| 10 | 2.5 | 574 | 296 | |
| 11 | 2.0 | 1.5 | 466 | 237 |
| 12 | 2.0 | 642 | 321 | |
| 13 | 2.5 | 629 | 329 | |
| 14 | 5.0 | 2.0 | 543 | 290 |
| 15 | 2.5 | 571 | 303 | |
| 16 | 5.0 | 432 | 232 | |
Fig. 1 shows the dependence of the rate of H2 and O2 evolution under UV irradiation (λ > 300 nm) on the calcination temperature after impregnation with 1.5 wt% Cu and 2.0 wt% Cr. The rates of H2 and O2 evolution increased significantly with increasing calcination temperature to a maximum at 623 K, beyond which the activity of the samples dropped sharply.
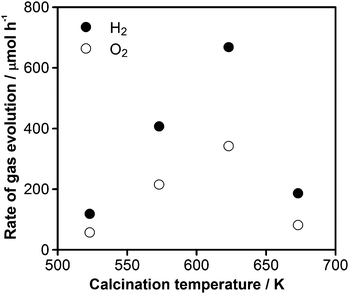 | ||
| Fig. 1 Dependence of the photocatalytic activity for overall water splitting on 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO after calcination at various temperatures. Reaction conditions: catalyst, 0.3 g; distilled water, 400 mL; light source, high-pressure mercury lamp (450 W); reaction vessel, Pyrex inner-irradiation-type; irradiation wavelength, λ > 300 nm. | ||
A typical time course of overall water splitting under visible light (λ > 400 nm) over the optimized catalyst (1.5 wt% Cu and 2.0 wt% Cr calcined at 623 K) is shown in Fig. 2. Although the rate of O2 evolution observed at the initial stage of the reaction (∼5 h) was larger than that expected from the stoichiometry, the gas evolution behavior gradually became stoichiometric (H2/O2 = 2), producing approximately 50 μmol h−1 H2 and 25 μmol h−1 O2, respectively. In contrast, a catalyst modified with either 1.5 wt% Cu or 2.0 wt% Cr did not produce any appreciable gas evolution. Therefore, modification of GaN:ZnO with proper amounts of Cu and Cr species and an appropriate final calcination temperature are both essential to enhance the water splitting rate.
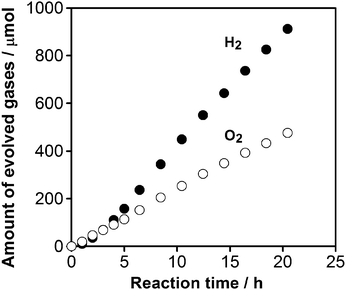 | ||
| Fig. 2 Time course of overall water splitting under visible light over 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO (calcined at 623 K). Reaction conditions: catalyst, 0.3 g; distilled water, 400 mL; light source, high-pressure mercury lamp (450 W); reaction vessel, Pyrex inner-irradiation-type with an NaNO2 solution (2 M) filter; irradiation wavelength, λ > 400 nm. | ||
TEM observation and EDS measurements
HR-TEM images of GaN:ZnO with 1.5 wt% Cu and 2.0 wt% Cr compositions after calcination at 623 K (the optimized catalyst) are shown in Fig. 3. On the larger particles of GaN:ZnO, smaller nanoparticles having several tens of nanometers in size with some aggregations were observed. Some of the nanoparticles produced lattice fringes, although these were not very clear, indicating that the loaded nanoparticles were crystallized to some extent. As shown in Table 2, the results of combined TEM-EDS analyses for this sample indicated that these nanoparticles contained both Cu and Cr, suggesting that the loaded nanoparticles were mixed oxides of Cu and Cr. Furthermore, these nanoparticles tended to lie between GaN:ZnO particles (part (c) of Fig. 3); in other words, the dispersion of the Cu–Cr nanoparticles was poor. However, the presence of the loaded Cu–Cr nanoparticles on GaN:ZnO could not be identified by X-ray diffraction analyses, presumably due to the low concentration and/or incomplete crystallinity (Figure S1†).
 | ||
| Fig. 3
HR-TEM images of GaN:ZnO loaded with 1.5 wt% Cu and 2.0 wt% Cr followed by calcination at 623 K. Spots of (a) were examined for reference, and almost no signals from Cu and/or Cr could be detected. | ||
:ZnO (calcined at 623 K)
XAFS measurements
The valence state of the loaded Cu and Cr species on GaN:ZnO was investigated by XAFS. Fig. 4A shows the Cu–K edge X-ray absorption near-edge structure (XANES) spectra for Cu-loaded GaN:ZnO with and without Cr co-loading. The catalyst modified with only Cu (calcined at 623 K) had a spectral shape similar to the CuO reference, judging from the pre-edge feature. This implies that the Cu species on GaN:ZnO were loaded as CuO, in good agreement with a previous report by Sayama et al., who demonstrated that impregnated Cu(NO3)2 on WO3 undergoes a structural transformation into CuO at 573 K.12 When Cr was co-loaded with Cu onto GaN:ZnO, the pre-edge structure of CuO disappeared, suggesting that the Cu species interacted with the Cr species on GaN:ZnO, in good agreement with the TEM-EDS results (Table 2). Fig. 4B shows the Cu–K edge XANES spectra for samples with 1.5 wt% Cu and 2.0 wt% Cr after calcination at various temperatures. The absorption edges of the prepared catalysts were located between those of Cu(NO3)2 and CuCr2O4, and the position shifted to lower photon energy with increasing calcination temperature. This indicates that the structure of the loaded Cu species became closer to CuCr2O4 than Cu(NO3)2 as the calcination temperature was increased. Fig. 4C shows Fourier transforms of the k3-weighted Cu–K edge EXAFS spectra for catalysts calcined at various temperatures. The spectral shapes of catalysts calcined below 573 K were somewhat similar. Starting at 623 K, however, a new peak appeared at 2.3–3.5 Å, and this peak was more pronounced at 673 K. Since Cu and Cr species interacted with each other in the loaded nanoparticles on GaN:ZnO, this spectral feature implies the generation of a Cu–(O)–Cr configuration and the aggregation of loaded nanoparticles, although the 673 K sample did not completely correspond to the bulk CuCr2O4 reference.
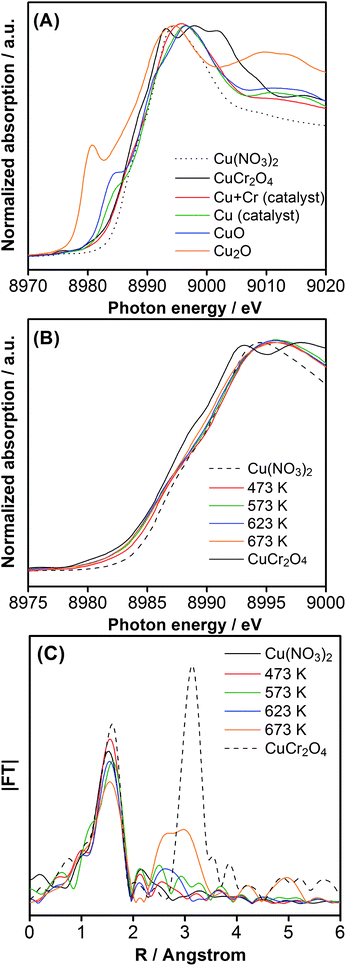 | ||
| Fig. 4 (A) Cu–K edge XANES spectra for GaN:ZnO catalysts co-loaded with 1.5 wt% Cu and 2.0 wt% Cr, and loaded with only 1.5 wt% Cu, with a common calcination temperature of 623 K. (B) Cu–K edge XANES spectra for 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO calcined at various temperatures. (C) Fourier transforms of k3-weighted Cu–K edge EXAFS spectra for 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO calcined at various temperatures. | ||
Fig. 5A shows the Cr–K edge XANES spectra for Cr-loaded GaN:ZnO with and without Cu co-loading. The catalyst modified with only Cr (calcined at 623 K) had a pre-edge peak assigned to hexavalent Cr, and a spectral feature similar to that of trivalent Cr around ca. 6020 eV. Therefore, Cr species on GaN:ZnO existed as a mixture of trivalent and hexavalent species. The Cu–Cr co-loaded catalyst exhibited a similar absorption profile, but the contribution of Cr(VI) species was more pronounced. However, the spectra of both catalysts were dissimilar to those of Cr(NO3)3 and CuCr2O4. Fig. 5B shows Cr–K edge XANES spectra of samples with 1.5 wt% Cu and 2.0 wt% Cr after calcination at various temperatures. The spectra of the catalysts calcined below 573 K were identical to that of CrO3, indicating that the valence state of the Cr species in these catalysts was very close to hexavalent. This result is reasonable, since the thermal decomposition of Cr(VI)-oxide begins at 573 K to yield Cr2O3.13 The spectral feature derived from Cr(III)-oxide begins to appear at 623 K, while the intensity of the Cr(VI) pre-edge peak diminishes. At 673 K, the spectrum becomes closer to the CuCr2O4 reference, with a further reduction of the Cr(VI) pre-edge peak, judging from the spectral shapes around the main absorption and 6020 eV-region. This structural change is also evident from the change in the FT of the EXAFS spectra, as shown in Fig. 5C. The spectrum of the catalyst calcined at 473 K was similar to that of CrO3, judging from the peak positions of the first and second shells. With increasing calcination temperature, however, the spectral shape of the catalyst samples began to undergo a change to that of CuCr2O4. At 673 K, the intensity of the peak appearing at 2.5 Å, assignable to the Cr–(O)–Cu or Cr (2.5 Å) shell configuration, increased significantly, indicating that the Cu–Cr mixed-oxide nanoparticles on GaN:ZnO were aggregated and the Cr-species were close to those of the bulk CuCrO4 reference after calcination at 673 K.
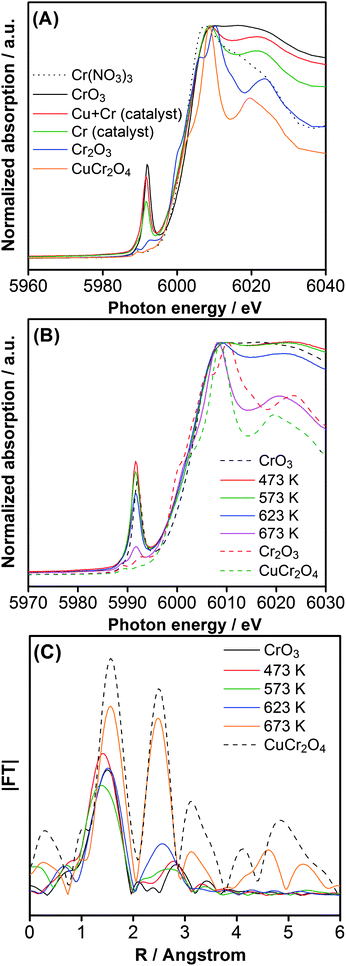 | ||
| Fig. 5 (A) Cr–K edge XANES spectra for GaN:ZnO catalysts co-loaded with 1.5 wt% Cu and 2.0 wt% Cr, and loaded with only 2.0 wt% Cr, with a common calcination temperature of 623 K. (B) Cr–K edge XANES spectra for 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO calcined at various temperatures. (C) Fourier transforms of k3-weighted Cr–K edge EXAFS spectra for 1.5 wt% Cu and 2.0 wt% Cr-loaded GaN:ZnO calcined at various temperatures. | ||
Considering the results of the photocatalytic reaction (Table 1 and Fig. 1), it appears that the aggregated state of the mixed-oxide nanoparticles was less effective as a cocatalyst for overall water splitting, which is consistent with the general trend of heterogeneous catalysis. Because the activity of a catalytic system is, in general, dependent on the surface area available for reaction, aggregation of nanoparticulate cocatalysts results in a drop in reaction rate, as exemplified by GaN:ZnO loaded with RuO24e or Rh2−yCryO3.5c Another possible explanation for the lower activity in higher loadings is an inner filter effect in which the loaded nanoparticulate cocatalysts prevent light absorption by the GaN:ZnO component.
On the basis of the above results, it can be concluded that Cu–Cr mixed-oxide nanoparticles with optimal composition and coverage are active cocatalysts for photocatalytic overall water splitting on GaN:ZnO. Nevertheless, an induction period was observed during visible-light-driven water splitting (Fig. 2). More specifically, the valence state of the Cu- and Cr-species may be reduced during the induction period, considering the less active water reduction activity.
XAFS measurements were therefore performed on a catalyst after 5 h of reaction (λ > 400 nm). As shown in Fig. 6A, the Cu–K edge spectrum remained nearly unchanged upon reaction, indicating that the Cu(II) state did not undergo reduction, even after the water splitting reaction. In the Cr–K edge spectrum, on the other hand, the pre-edge peak assigned to Cr(VI) disappeared completely, suggesting that Cr(VI) species existing on the surface of GaN:ZnO before reaction was reduced to the Cr(III) state. Presumably, the remaining Cr(VI) species in the as-prepared catalyst dissolved into the reactant solution, undergoing photoreduction during overall water splitting to give Cr(III) oxide on Cu(II)–Cr(III) mixed-oxide nanoparticles,6 as has been observed in GaN:ZnO modified with Rh(III)–Cr(III) mixed-oxide nanoparticles.5b Therefore, the active valence states of Cu and Cr in the mixed-oxide nanoparticles for photocatalytic water splitting are Cu(II) and Cr(III), respectively. However, HR-TEM observations revealed that the reacted catalyst exhibited no significant difference in morphology from that of the unreacted catalyst (Figure S2†).
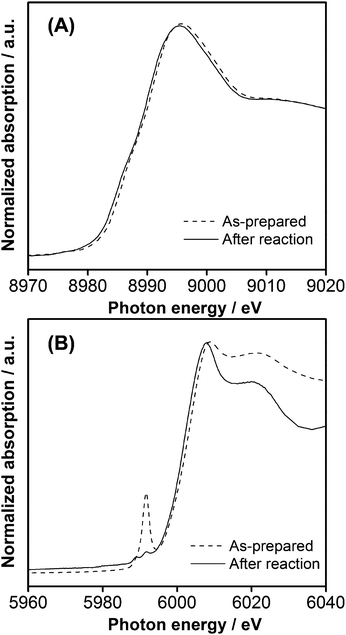 | ||
| Fig. 6 (A) Cu–K and (B) Cr–K edge XANES spectra for 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO (calcined at 623 K) before and after reaction (5 h, λ > 400 nm). | ||
Electrochemical measurements
On the basis of the XAFS measurements, it was concluded that the generation of Cu(II)–Cr(III) mixed-oxide (referred to as CuCrOx hereafter for simplicity) nanoparticles with optimal composition and coverage contributed to the enhancement of the water splitting rate over GaN:ZnO. However, the promotional effect of the Cr component on the water splitting rate remained unclear. Sayama et al. have reported that CuO on WO3 collects the conduction band electrons of WO3, thereby promoting the oxygen reduction reaction during the decomposition of organic substrates.15 The photoreduction of O2 is an essential step to promote the decomposition of organic substrates, but is an undesirable reverse reaction of overall water splitting.5b,6d,14 Thus, one plausible reason for the improvement of catalytic activity by Cr co-loading is that the paired Cr component suppresses the photoreduction of O2 catalyzed by the Cu-oxide species loaded on the GaN:ZnO.
The behavior of CuCrOx/GaN:ZnO with respect to the reduction of O2 was therefore investigated using an electrochemical cell by monitoring the cathodic response under dark conditions with continuous gas bubbling. As shown in Fig. 7, an appreciable cathodic current was observed when the CuO/GaN:ZnO electrode was employed in the presence of O2, and this current increased with increasing negative potential. On the other hand, the CuCrOx/GaN:ZnO electrode generated a very small cathodic current in the potential range examined here. It was also confirmed that there was no cathodic current for any of the prepared electrodes in the absence of O2 gas. These results suggest that CuCrOx nanoparticles on GaN:ZnO are largely insensitive to the reduction of O2; in other words, the addition of Cr to CuO/GaN:ZnO has a positive effect on the suppression of O2 photoreduction. It was also confirmed that nanoparticulate CuCrOx on GaN:ZnO does not catalyze water formation from H2 and O2 (Figure S3†), another backward reaction in overall water splitting.
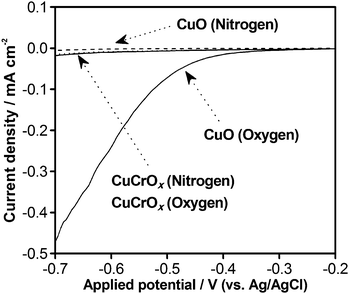 | ||
| Fig. 7 Current–voltage curves for porous CuCrOx/GaN:ZnO and CuO/GaN:ZnO electrodes under dark conditions in an aqueous solution containing 0.1 M Na2SO4 with N2 or O2 bubbling. Scan rate: 5 mV s−1. | ||
Suppression of backward reaction by introducing chromium component
To obtain further insight into the photoreduction of O2 over CuCrOx/GaN:ZnO catalyst, overall water splitting reactions were performed in the presence of O2. Fig. 8 shows the time course of H2 evolution in visible-light-driven overall water splitting, where 30 kPa of O2 gas was intentionally introduced before irradiation. The rate of H2 evolution was approximately 50 μmol h−1, which is identical to that observed in overall water splitting (Fig. 2). This result clearly indicates that the photoreduction of O2 on CuCrOx/GaN:ZnO during the overall water splitting reaction is very slow, in good agreement with the results of electrochemical measurements (Fig. 7).
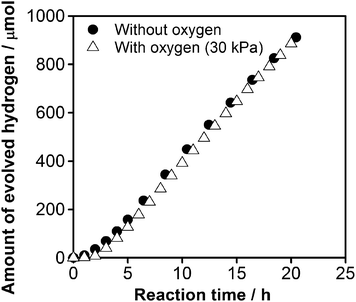 | ||
| Fig. 8 Time course of overall water splitting under visible light over 1.5 wt% Cu and 2.0 wt% Cr loaded GaN:ZnO (calcined at 623 K). Reaction conditions: catalyst, 0.3 g; distilled water, 400 mL; light source, high-pressure mercury lamp (450 W); reaction vessel, Pyrex inner-irradiation-type with a NaNO2 solution (2 M) filter; irradiation wavelength, λ > 400 nm. | ||
As suggested by the electrochemical measurement results, the inactivity of CuO/GaN:ZnO for water splitting was most likely due to the photoreduction of O2 catalyzed by the loaded CuO. Interestingly, however, H2 and O2 evolution over CuO/GaN:ZnO were both observed when the reaction was conducted in an aqueous K2CrO4 solution (Figure S4†).15 At the initial stage of the reaction, the ratio of H2/O2 evolution was smaller than that expected from stoichiometry, indicating that Cr(VI) ions in the reactant solution were reduced by photogenerated electrons from the conduction band of GaN:ZnO, thereby lowering the water reduction efficiency.6b,d This tendency is similar to that observed in overall water splitting using CuCrOx/GaN:ZnO, as shown in Fig. 2. After filtration of the above reactant solution, the resulting sample was again tested for overall water splitting without K2CrO4 under visible light. It was found that approximately 12.0 μmol h−1 H2 and 6.0 μmol h−1 O2 were produced at the steady state (Figure S5†).
The K2CrO4 treatment shown above results in the formation of a Cr2O3-shell on the cocatalyst component through photoreduction,6 which prevents the backward reaction over the cocatalyst (water formation from H2 and O2 and photoreduction of O2); specifically, the amorphous Cr2O3 shell-layer is permeable to protons and the evolved H2 molecules, but not to O2.16 Therefore, the appreciable H2 and O2 evolution observed both in the illuminated K2CrO4 and after reuse of the treated catalyst (Figures S4 and S5†) was likely to be due to the formation of a protective Cr2O3 layer that suppressed the backward reactions; in this case, the photoreduction of O2 that would occur on CuO. In our earlier work, Rh2O3-loaded GaN:ZnO was shown to exhibit a behavior quite similar to the present CuO/GaN:ZnO.6d
Based on these results, it is reasonable to conclude that CuO on GaN:ZnO can accept electrons from GaN:ZnO and host active sites for H2 evolution if the undesirable backward reaction is effectively suppressed. Similarly, CuCrOx on GaN:ZnO is likely to host H2 evolution sites, although the details of how the backward reaction can be suppressed on a molecular level and how H2 evolution occurs on the CuCrOx cocatalyst during water splitting using GaN:ZnO remain unclear. We are currently investigating the reaction mechanism through a variety of spectroscopic techniques.
Conclusions
A mixed oxide of Cu and Cr was examined as a noble-metal-free cocatalyst for photocatalytic overall water splitting with a GaN:ZnO solid solution. The water splitting rate was maximized by adding appropriate amounts of both Cu and Cr to promote the forward reaction while avoiding excess coverage of GaN:ZnO, and by choosing the proper calcination temperature after impregnation of the Cu and Cr precursors. Nanoparticles of divalent Cu and trivalent Cr mixed-oxide with an optimal composition and distribution, as determined by the preparation conditions, were demonstrated to act as active cocatalysts for photocatalytic overall water splitting, and were largely resistant to the undesirable backward reactions of water formation from a H2/O2 mixture and the photoreduction of O2.
The photocatalytic activity of the optimized CuCrOx/GaN:ZnO for overall water splitting was at most 25–30% of that obtained using a similarly optimized catalyst loaded with Rh2−yCryO3, which is the most effective currently-known cocatalyst. This is at least in part due to the poorly dispersed CuCrOx nanoparticles on GaN:ZnO, which lowered the active surface area for water reduction. Nevertheless, the present result is encouraging, because the price of Cu is approximately three orders of magnitude lower than that of Rh. As mentioned in the Introduction, the lower cost of a catalyst component would become advantageous, when one considers a large-scale application. In addition, it is possible to improve the dispersion of nanoparticulate cocatalysts on a photocatalyst by refining the loading method.17 Efforts are currently underway to do so, as well as to elucidate the detailed reaction mechanism.
Acknowledgements
The authors thank the staff of Mitsubishi Chemicals Co. for TEM observation and EDS analysis. This work was supported by the Research and Development in a New Interdisciplinary Field Based on Nanotechnology and Materials Science program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and the KAITEKI Institute, Inc. One of the authors (K. M.) thanks Nippon Sheet Glass Foundation for Materials Science and Engineering.Notes and references
- (a) P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834 CrossRef CAS; (b) M. Pagliaro, A. G. Konstandopoulos, R. Ciriminna and G. Palmisano, Energy Environ. Sci., 2010, 3, 279 RSC; (c) R. Schlögl, ChemSusChem, 2010, 3, 209 CrossRef.
- (a) J. S. Lee, Catal. Surv. Asia, 2005, 9, 217 CrossRef CAS; (b) K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CrossRef CAS; (c) Y. Inoue, Energy Environ. Sci., 2009, 2, 364 RSC; (d) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (e) W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966 CrossRef CAS; (f) K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 Search PubMed.
- (a) K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543 RSC; (b) H. G. Kim, D. W. Hwang, J. Kim, Y. G. Kim and J. S. Lee, Chem. Commun., 1999, 1077 RSC; (c) H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS; (d) S. Ikeda, T. Itani, K. Nango and M. Matsumura, Catal. Lett., 2004, 98, 229 CrossRef CAS; (e) T. Yanagida, Y. Sakata and H. Imamura, Chem. Lett., 2004, 33, 726 CrossRef CAS.
- (a) Y. Inoue, T. Kubokawa and K. Sato, J. Chem. Soc., Chem. Commun., 1990, 1298 RSC; (b) Y. Inoue, T. Niiyama, Y. Asai and K. Sato, J. Chem. Soc., Chem. Commun., 1992, 579 RSC; (c) J. Sato, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2001, 105, 6061 CrossRef CAS; (d) K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS; (e) K. Teramura, K. Maeda, T. Saito, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2005, 109, 21915 CrossRef CAS.
- (a) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS; (b) K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107 CrossRef CAS; (c) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753 CrossRef CAS; (d) K. Maeda, D. Lu, K. Teramura and K. Domen, J. Mater. Chem., 2008, 18, 3539 RSC; (e) K. Maeda, D. Lu, K. Teramura and K. Domen, Energy Environ. Sci., 2010, 3, 471 RSC.
- (a) K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS; (b) K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554 CrossRef CAS; (c) N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106 RSC; (d) K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750 CrossRef CAS.
- (a) X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176 CrossRef CAS; (b) X. Zong, G. Wu, H. Yan, G. Ma, J. Shi, F. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114, 1963 CrossRef CAS.
- J. S. Jang, D. J. Ham, N. Lakshminarasimhan, W. Choi and J. S. Lee, Appl. Catal., A, 2008, 346, 149 CrossRef CAS.
- (a) M. Tabata, K. Maeda, T. Ishihara, T. Minegishi, T. Takata and K. Domen, J. Phys. Chem. C, 2010, 114, 11215 CrossRef CAS; (b) W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631 RSC.
- (a) M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS; (b) F. Jiao and H. M. Frei, Chem. Commun., 2010, 46, 2397 RSC.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303 CrossRef CAS.
- T. Arai, M. Horiguchi, M. Yanagida, T. Gunji, H. Sugihara and K. Sayama, J. Phys. Chem. C, 2009, 113, 6602 CrossRef CAS.
- D. R. Lide, Handbook of Chemistry and Physics, 83rd ed.; CRC Press: Boca Raton, FL, 2002 Search PubMed.
- (a) K. Domen, A. Kudo and T. Onishi, J. Catal., 1986, 102, 92 CrossRef CAS; (b) K. Sayama and H. Arakawa, J. Chem. Soc., Faraday Trans., 1997, 93, 1647 RSC; (c) K. Sayama and H. Arakawa, J. Photochem. Photobiol., A, 1996, 94, 67 CrossRef CAS.
- The reaction was carried out in a similar manner, but with an aqueous solution containing K2CrO4 (5.0 wt% Crvs.CuO/GaN:ZnO). See the caption of Figure S4 in ESI†.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151 CrossRef CAS.
- (a) N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106 RSC; (b) K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details, TEM images and reaction data. See DOI: 10.1039/c1sc00177a |
| This journal is © The Royal Society of Chemistry 2011 |