Surface state during activation and reaction of high-performing multi-metallic alkyne hydrogenation catalysts†
Blaise
Bridier
a,
Javier
Pérez-Ramírez
*a,
Axel
Knop-Gericke
b,
Robert
Schlögl
b and
Detre
Teschner
*b
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, HCI E 125, Wolfgang-Pauli-Strasse 10, CH-8093, Zurich, Switzerland. E-mail: jpr@chem.ethz.ch; Fax: +41 44 633 14 05; Tel: +41 44 633 71 20
bFritz-Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, Faradayweg 4–6, Berlin, D-14195, Germany. E-mail: teschner@fhi-berlin.mpg.de; Fax: +49 30 8413 4676; Tel: +49 30 8413 5408
First published on 13th May 2011
Abstract
In partial hydrogenation of highly unsaturated compounds, high-performance heterogeneous catalysts usually consist of multi-metallic systems providing enhanced selectivity. These materials often undergo complex segregation phenomena and to understand their function, a surface-sensitive in situ methodology is crucial. Recently, we reported a novel family of ternary Cu–Ni–Fe catalysts for propyne hydrogenation with exceptional selectivity to propene. Herein, we detail our study on the surface composition and electronic state of two representative samples (Cu2.75Ni0.25Fe and Cu3Fe) using in situ X-ray photoelectron (XPS) and X-ray absorption (XAS) spectroscopies. Surface segregation phenomena during activation of the catalyst precursors (calcination and reduction) and hydrogenation reaction were evaluated. The multiple functions of nickel in the catalyst, which account for the extraordinary alkene selectivity, are unravelled.
Introduction
Heterogeneous catalysts are frequently made of multiple components forming the catalytically active and/or promoter phases. For example, current research in the field of partial hydrogenation of highly unsaturated compounds is focusing on the use of alloys in order to improve the catalytic performance.1–4 Although alloys, in comparison to the monometallic analogues, often increase activity and/or selectivity, solid-solution type alloys are in principle not desirable since segregation of constituent elements may turn the catalyst into an undesirable state. The surface of the solid material is vital for the catalytic process and therefore any evolution of its nature may cause drastic changes in activity, selectivity, and/or lifetime. Our recent work in the topic of partial alkyne hydrogenation on Ni3Al and Cu3Al hydrotalcite-derived catalysts showed that upon calcination and reduction treatment, the catalysts suffered from a strong aluminium surface enrichment. The aluminium tends to shield the catalysts, thereby reducing the fraction of exposed hydrogenation sites.5,6 This phenomenon highlights the importance of surface analyses in order to unravel the true state and composition of surfaces during reaction, so that catalysts can be optimally designed for their particular application.During the last 20 years, researchers have developed numerous in situ characterisation methods to provide a better understanding of the surface structure and valence state of catalysts under their operation.7–11In situX-ray photoelectron spectroscopy (XPS) in the mbar pressure range is an excellent tool for studying the electronic structure of the catalyst surface under reaction conditions.12 It has provided numerous insights into the surface and subsurface (carbides, hydrides) chemistry of the widely used Pd-based catalysts in alkyne hydrogenation.13,14 Moreover, Somorjai et al.15 have shown that alloy nanoparticles undergo remarkable surface changes in composition and chemical state in response to the applied gaseous atmosphere. Complementarily, computational studies from Nørskov et al.16–19 have predicted segregation phenomena in pairs of metal. Surface segregation was shown to depend on the crystal structure, particle size, surface orientation, and adsorbates from the gas phase. Hence, the surface and near-surface region of catalysts are dynamic and reflect always the actual ambient (gas composition, pressure and temperature). Therefore, in situXPS stands as a key technique to derive robust relationships between the surface structure and catalytic performance.
Recently, we identified a novel family of ternary Cu–Ni–Fe catalysts for the gas-phase partial hydrogenation of propyne with alkene selectivity approaching 100%.20 The superior performance of the ternary systems was tentatively attributed to the cooperating Ni and Fe promotion of the Cu-based catalysts. Since the surface state of these materials hold the key of their exceptional selectivity, and this type of complex ternary catalyst is expected to exhibit surface segregation phenomena, we have carried out in situXPS and in situX-ray absorption spectroscopy (XAS) experiments. These methods enable us to investigate the surface state of these samples under hydrogenation conditions. We have compared the binary Cu3Fe and ternary Cu2.75Ni0.25Fe catalysts since a slight addition of Ni to the binary system is optimal to attain a remarkable effect on the product distribution.20 Whereas Cu3Fe showed equivalent propene selectivity (70%) as its aluminium counterpart,6 the Cu2.75Ni0.25Fe catalyst under the same condition gave superior propene selectivity up to 97% with only 3% of oligomers. None of the catalysts produced propane.
In this Edge Article, we confront bulk and in situ surface analyses in order to gain insights into the chemical state of the surface and composition changes responsible of the outstanding performance of Cu2.75Ni0.25Fe in partial alkyne hydrogenation. We investigate surface segregation effects upon calcination, reduction, and reaction. Multiple promoting effects of nickel in the ternary catalyst have been identified and discussed.
Results and discussion
Prior to their use in propyne hydrogenation, both Cu3Fe and Cu2.75Ni0.25Fe samples were investigated in their calcined and ex situ reduced states. The reader can find detailed experimental descriptions in the Electronic Supplementary Information (ESI†). The chemical state of the catalysts is summarised in Table 1, whereas the atomic fraction of the main surface constituents (inelastic mean free path: 9 Å)21 is depicted in Fig. 1. Based on XRD,20 the bulk of the binary Cu3Fe sample in the calcined state consists of tenorite (CuO) as the main phase and CuFe2O4 spinel as secondary phase. On the other hand, no spinel was identified on the surface by XAS and, instead, Fe2O3 (hematite) was observed. This difference was found to persist with Cu2.75Ni0.25Fe (vide infra). Similarly to the penetration of copper ions in alumina reported by Bolt et al.,22 we can assume that copper segregates towards the bulk viaCuFe2O4 spinel formation at the expense of a surface enrichment of Fe2O3. Nonetheless, the surface iron concentration was practically identical to its bulk composition, which is in strong contrast to the case of Cu3Al,6 where aluminium strongly segregated to the surface (Fig. 1). Reduction of Cu3Fe gave rise to the formation of metallic Cu and Fe with some magnetite as detected by XRD in the reduced sample (Table 1). These phases are however buried by CuO and Fe2O3, since the phases detected by surface-sensitive XPS and XAS techniques in the reduced sample seem to be identical to those in the calcined precursor. This was, apparently, due to surface re-oxidation while the sample was carried to the synchrotron. When replacing part of the Cu by Ni, both bulk and surface of the calcined Cu2.75Ni0.25Fe seemed to be basically unaffected. In the bulk, no Ni-based phase was observed, and Ni2+ cations are thus occupying metal sites in the spinel and CuO, the latter has been evidenced by lattice contraction in XRD.20 With XAS, however, we do witness some NiO at the surface (Fig. 2B, spectrum 1), but its concentration is rather small (40% of bulk Ni). As expected from computed surface segregation energies in transition-metal alloys,16 the pairs Cu–Ni and Fe–Ni, with Cu or Fe as host and Ni as solute, resulted upon calcination in moderate anti-segregation and in strong segregation, respectively. Following this reasoning, we speculate that Fe–Ni segregation at the surface leads to the appearance of NiO. Although hematite was slightly enriched at the surface, its magnitude was significantly lower than the enrichment of aluminium, most likely as amorphous Al2O3, in Cu2.75Ni0.25Al (Fig. 1, 1st column).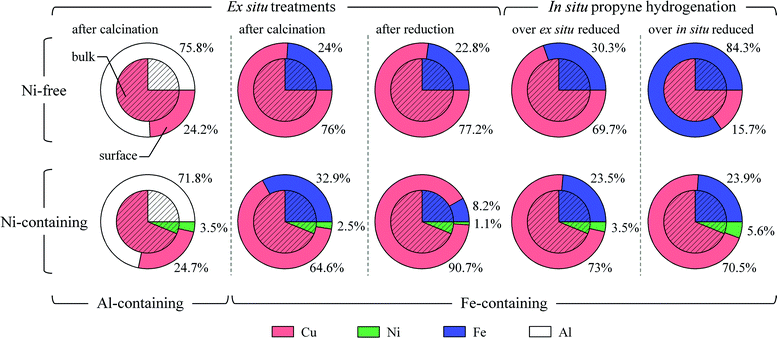 | ||
| Fig. 1 Surface (shell) and bulk (core) composition according to XPS analysis, and the theoretical bulk ratio of the Cu3Fe and Cu2.75Ni0.25Fe samples after calcination, ex situreduction, and under propyne hydrogenation (after ex situ or in situreduction). Conditions: Calcined Cu3Fe and Cu2.75Ni0.25Fe were measured under O2 at 573 and 523 K, respectively, whereas their ex situ pre-reduced states were acquired under UHV at room temperature. Propyne hydrogenation was performed at 523 K and 1 mbar using a reaction mixture of H2/C3H4 = 3. For comparison, XPS results of the calcined Cu3Al and Cu2.75Ni0.25Al are included (1st column). | ||
| Sample | Surface analysis (XAS) | Bulk analysis (XRD) | |||||
|---|---|---|---|---|---|---|---|
| Atm./Temp. | Cu-phase | Fe-phase | Ni-phase | Cu-phase | Fe-phase | Ni-phase | |
| a R-mix refers to ‘reaction mixture’. Other conditions: H2/C3H4 = 3, P = 1 mbar, and F = 15 cm3 STP min−1. | |||||||
| Cu3Fe calcined | O2/573 K | CuO | Fe2O3 | — | CuO | CuFe2O4 spinel | — |
| R-mixa/523 K | Cu | Fe/Fe3O4/Fe2O3 (5/89/6) | |||||
| Cu3Fe reduced | He/RT | CuO | Fe2O3 | — | Cu | Fe/Fe3O4 | — |
| R-mix/523 K | Cu | Fe/Fe3O4/Fe2O3 (12/63/25) | |||||
| Cu2.75Ni0.25Fe calcined | O2/523 K | CuO | Fe2O3 | NiO | CuO | CuxNi1−xFe2O4 spinel | Not detected |
| R-mix/523 K | Cu | Fe/Fe3O4/Fe2O3 (72/14.5/13.5) | Ni/NiC | ||||
| Cu2.75Ni0.25Fe reduced | O2/RT | Cu/Cu2O/CuO (46/43/11) | Fe/Fe3O4/Fe2O3 (53/6/41) | Ni/NiO (92/8) | Cu | Fe/Fe3O4 | Ni |
| R-mix/523 K | Cu | Fe/Fe3O4/Fe2O3 (68/11/21) | Ni/NiC | ||||
Unlike Cu3Fe, both the composition and chemical state of Cu2.75Ni0.25Fe have been modified at the surface after ex situreduction. Cu is massively enriched at the surface from 64.6% up to 90.7% at the expense of both Fe and Ni. Copper was in a mixed state with metallic Cu (∼46%), Cu2O (∼43%) and some CuO (∼11%), as shown in Fig. 2A (spectrum 2). It should be noted that the composition of a constituent in XAS is proportional to the edge jump and not to the height of strong resonances (e.g.CuO). According to the iron and nickel L3-edges, iron is in the state of mainly Fe (∼53%) and Fe2O3 (∼41%) with a minor Fe3O4 (∼6%) contribution, whereas nickel is predominantly (∼92%) metallic. Thus comparing the two samples, we can conclude that Ni enhances the reduction of Cu and Fe on the surface, which is in agreement with our previous bulk H2-TPR study,20 as well as hinders their re-oxidation. Furthermore, Ni promotes surface enrichment of metallic Cu in the catalyst under a reductive atmosphere. These results provide valuable understanding on the role(s) of Ni as promoter in the ternary catalyst. It should not be merely regarded as a H-coverage enhancer but also as a “driver” for copper from the bulk to the surface during reduction. Despite segregation phenomena during activation, strong interaction in the bulk was evidenced in the XRD pattern of the reduced samples by the shift of the main Cu(111) reflection to higher diffraction angles (Fig. ESI 1†). Despite the low nickel content in the ternary sample, the additional shift from Cu3Fe to Cu2.75Ni0.25Fe is in line with the predicted anti-segregation of the Ni–Cu system as Ni is well dispersed in the bulk and further affects the Cu structure.16
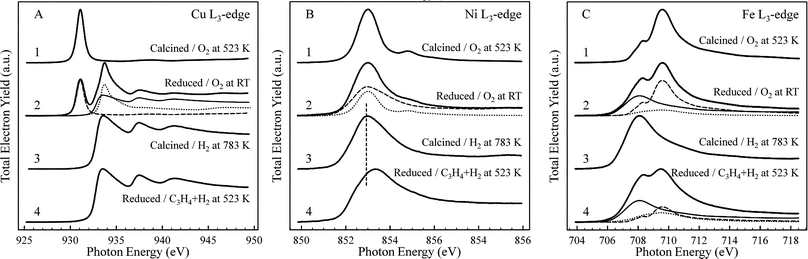 | ||
| Fig. 2 Cu (A), Ni (B) and Fe (C) L3-edge of the calcined and ex situ reduced Cu2.75Ni0.25Fe at different conditions. The labels indicate the state of the catalyst and the conditions (atmosphere and temperature) during spectroscopic analysis. 1: calcined/0.2 mbar O2 at 523 K; 2: reduced/0.2 mbar O2 at RT; 3: calcined/1 mbar H2 at 783 K; 4: reduced/1 mbar C3H4 + H2 at 523 K. Fit components: in (A) Cu L3: Cu (full), Cu2O (dotted) and CuO (dashed); in (B) Ni L3: Ni (dashed) and NiO (dotted); in (C): Fe L3: Fe (full), Fe3O4 (dotted) and Fe2O3 (dashed). | ||
To illustrate the surface re-oxidation of the ex situ reduced samples during transfer to the synchrotron, we have carried out spectroscopic analyses during in situreduction of the calcined samples. As Cu3Fe reduces at slightly higher temperature than Cu2.75Ni0.25Fe,20 we have chosen reduction temperatures of 855 and 783 K, respectively. At these conditions, both samples were fully reduced in the investigated information depth, as shown by the example of Cu2.75Ni0.25Fe in Fig. 2A–C, spectra 3. The X-ray absorption spectra of all constituents could be well described by their metallic states.23,24
As we have reported recently,20 both copper-based catalysts perform well in partial propyne hydrogenation, Cu2.75Ni0.25Fe being exceptionally selective even at a full degree of alkyne conversion. Table 2 compiles the hydrogenation performance during XPS/XAS measurement and in flow tests at 1 bar (testing procedure detailed in ESI†).
With a propene selectivity of 97%, Cu2.75Ni0.25Fe is one of the most efficient catalysts ever reported for this reaction. During in situXPS, both catalysts showed a low degree of alkyne conversion, which however was expected considering the large XPS cell volume (5 L) with a small 5 mm diameter catalyst pellet inside, and the relatively high gas-flow rate. Clearly, the XPS chamber was not constructed to reach significant conversion levels, but rather to elucidate the effect of different gas-phase conditions on the surface state. Nevertheless, the low activity was combined with high selectivity, which is representative at such a low conversion, and thus, the lack of pressure dependence in the selectivity strengthens the bridge between the low- and high-pressure structure–activity relationship and makes it possible to compare the state of the catalysts under these conditions.
During hydrogenation at 1 mbar, copper was metallic in both ex situ reduced samples; however the iron state was fairly different. In Cu3Fe, we observed mainly (∼63%) Fe3O4 with less (∼25%) Fe2O3 and a minority of (∼12%) Fe, hence full reduction of Fe2O3 to Fe was not obtained, whereas in Cu2.75Ni0.25Fe, ∼68% of the iron was metallic with the remaining ∼21% Fe2O3 and ∼11% Fe3O4. The state of nickel was somewhat puzzling as we observed an unambiguous shift of the resonance maximum (Fig. 2B, spectrum 4) from that of metallic Ni, and the shape of the spectrum is also clearly modified. We can exclude oxide formation, as evidenced by the comparison to NiO (Fig. 2B, spectrum 1), but we might speculate that Ni was strongly interacting with carbon from the alkyne feed. This assignment is supported by the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2p spectrum under alkyne hydrogenation (see Fig. ESI 2†), indicating a reduced asymmetry of the peak shape, as well as a +0.3 eV binding energy shift compared to the peak in pure H2 (852.8 eV). This is in line with data of Zdansky et al.25 over Ni(100) reporting Ni2p binding energies of 853.2 eV for surface-carbon modified Ni and 853.0 eV for Ni with subsurface carbon. It is well known that nickel can form carbides,26,27 and thus we tentatively assign the XAS signature as “carbidic”. The reader should note that the C1s signal is not informative here, as the carbidic species is an extreme minority component of the C1s spectrum as can be expected from the overall low Ni content, and the strong overlap of this minority species with the main C1s signal makes the carbide identification unreliable. As shown by Fig. 1 (4th column), nickel was still depleted on the surface, but much less so than in pure hydrogen, with the overall atomic fraction approaching the bulk composition. This effect can be attributed to the preferred interaction of Ni and carbon, and corroborates the XAS assignment of Ni. The carbon aided segregation of Ni is not a particular feature of our catalyst as adsorbate induced segregation has been reported for other alloys.15,18,28 The modification of Ni due to the carbide formation is in agreement with the evolution of the propene and propane selectivity during time-on-stream at 1 bar (Fig. ESI 3†). The increase in propene selectivity at the beginning of the reaction has been attributed to a beneficial carbon deposition that slightly hindered the hydrogen activation ability of Ni in order to enhance its selectivity.5,27
2p spectrum under alkyne hydrogenation (see Fig. ESI 2†), indicating a reduced asymmetry of the peak shape, as well as a +0.3 eV binding energy shift compared to the peak in pure H2 (852.8 eV). This is in line with data of Zdansky et al.25 over Ni(100) reporting Ni2p binding energies of 853.2 eV for surface-carbon modified Ni and 853.0 eV for Ni with subsurface carbon. It is well known that nickel can form carbides,26,27 and thus we tentatively assign the XAS signature as “carbidic”. The reader should note that the C1s signal is not informative here, as the carbidic species is an extreme minority component of the C1s spectrum as can be expected from the overall low Ni content, and the strong overlap of this minority species with the main C1s signal makes the carbide identification unreliable. As shown by Fig. 1 (4th column), nickel was still depleted on the surface, but much less so than in pure hydrogen, with the overall atomic fraction approaching the bulk composition. This effect can be attributed to the preferred interaction of Ni and carbon, and corroborates the XAS assignment of Ni. The carbon aided segregation of Ni is not a particular feature of our catalyst as adsorbate induced segregation has been reported for other alloys.15,18,28 The modification of Ni due to the carbide formation is in agreement with the evolution of the propene and propane selectivity during time-on-stream at 1 bar (Fig. ESI 3†). The increase in propene selectivity at the beginning of the reaction has been attributed to a beneficial carbon deposition that slightly hindered the hydrogen activation ability of Ni in order to enhance its selectivity.5,27
Similarly to Cu2.75Ni0.25Fe, the surface composition of Cu3Fe is not far from that of the bulk, with a slight excess of iron at the surface. Therefore, though both samples show segregation phenomena under reaction condition, the surface composition is surprisingly close to that of bulk. To understand the ordering and element distributions on the surface, we need to take into account the surface mixing energies calculated by Christensen et al.19 Since the ternary Cu–Fe–Ni system is unknown, we considered the binary Cu–Ni and Cu–Fe cases where Ni would mix with Cu in the surface, while Fe would separate and form islands. Thus the surface state would be mainly Cu, with some well dispersed Ni and monometallic Fe rafts, with little Fe–Cu intermixing. Additionally, some magnetite particles would decorate the surface and may serve as a “binder” between the primary alloy particles. The structural promotion of Fe should be highlighted in comparison to Al. Despite the strong segregation of the later, the use of Fe hindered Ni particle formation. This was clearly shown by the Ni-type behaviour of the Cu2.75Ni0.25Al catalyst during propyne hydrogenation (Fig. ESI 3†), as its activation period lasted 5 h from S(C3H6) = 20% to 87% compared to Cu2.75Ni0.25Fe, for which selectivity to propene increased from 86% to 97% within 0.5 h.
When investigating the hydrogen adsorption properties of CuNi alloy surfaces in UHV condition, Silvermann et al.29 observed a 50-fold decrease in the hydrogen adsorption probability over the Cu65Ni35 surface as compared to pure Ni(110). Nevertheless, the saturation coverage decreased by only a factor of 2.7, thus it was concluded that dissociative H2activation requires sites with numerous Ni neighbours. Since the Ni content of our Cu2.75Ni0.25Fe catalyst surface is almost an order of magnitude lower, and the activating period during propyne hydrogenation is insignificant, we can neglect the formation of multiple Ni ensembles. On the contrary, our recent calculations27 indicated that even a single Ni atom among Cu is sufficient to increase hydrogen activation compared to pure Cu and thus to enhance steady-state H-coverage. Based on the foregoing analyses, Scheme 1 illustrates the surface of Cu2.75Ni0.25Fe under reaction condition.
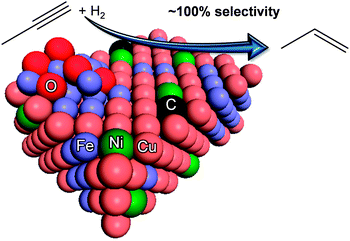 | ||
| Scheme 1 Schematic view of the surface of Cu2.75Ni0.25Fe under propyne hydrogenation based on in situ spectroscopic (XPS/XAS) analyses. | ||
To pinpoint the role of high-pressure reduction and that of Ni as hydrogen activator, we exposed the calcined samples after in situreduction to the same reaction experiment as detailed above. The elemental composition of this experiment is shown in Fig. 1 (5th column). Whereas the state and composition of Cu2.75Ni0.25Fe was very much comparable to the results with the ex situ reduced sample, huge compositional differences were observed with the nickel-free sample. Iron was preferentially enhanced at the surface with more than three-times higher Fe concentration than in the bulk. During H2 treatment, reduction of all surface oxide phases took place; however at the low pressure of 1 mbar, bulk reduction was kinetically hindered and thus not completed. While cooling down to 523 K, bulk oxygen still could diffuse to the surface, but reduction of Fe3O4 was no longer possible. Since copper is metallic at this condition, oxygen diffusion is necessarily coupled with iron segregation and thus, the composition gets reversed at the surface. It has been shown from XRD and H2-TPR20 that bulk Fe was not reduced entirely and therefore oxygen diffuses to the surface enriching the amount of surface Fe under reaction condition. On the other hand, the presence of Ni helps faster reduction and oxygen diffusion towards the surface. Furthermore, the Ni-carbon interaction allows nickel to participate in the surface and boosts its low surface concentration. Therefore these above phenomena can explain the depletion of Cu from the surface of the in situ reduced Cu2.75Ni0.25Fe under reaction condition.
Despite the surface composition of both ex situ reduced Cu3Fe and Cu2.75Ni0.25Fe catalysts during hydrogenation reaction being rather similar to the bulk, addition of even a small amount of Ni to the Cu-based catalyst will lead to different segregation events and induces the formation of diluted Cu–Ni ensembles with high enough hydrogen coverage to suppress oligomerisation but low enough to avoid over-hydrogenation. Such balance is crucial for highly selective surface reactions.
Conclusions
Based on in situXPS and XAS experiments, we have characterised the surface of binary Cu3Fe and ternary Cu2.75Ni0.25Fe catalysts used in partial alkyne hydrogenation. Despite the kinetic hindrance that occurs under low pressure (bulk reduction, low degree of conversion), the trend in the observed selectivity pattern as well as the role of Ni in defining surface properties strongly validates our approach. We show that the Cu3Fe is not significantly affected by calcination, reduction, or by the reaction media; and both copper and iron behave as if they are negligibly interacting. Nevertheless, a slight addition of Ni increases the inter-metallic interaction and yields a completely different picture. Ni not only enhances the H-coverage, as expected from the lower selectivity to oligomers, but drastically changes the surface composition. Ni enhances Cu and Fe reduction and hinders their re-oxidation that would favour Fe surface segregation. In addition, it promotes surface enrichment of metallic Cu in pure hydrogen. However, under reaction conditions, both Ni and Fe segregate back to the surface leading to a similar bulk–surface concentration. Therefore, the addition of Ni increases the cooperating effect of the ternary catalyst that leads to its superior alkene selectivity, compared to the binary counterparts. XPS in the mbar range has proved to be an excellent technique to unravel surface structure–performance relationship of a complex multi-metallic catalyst under operating conditions.Acknowledgements
Funding by ETH Zürich is acknowledged. We acknowledge the Helmholtz-Zentrum Berlin Electron storage ring BESSY II for providing synchrotron beamtime at the ISISS beamline.Notes and references
- B. Coq and F. Figueras, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef CAS.
- D. Mei, M. Neurock and C. Smith, J. Catal., 2009, 268, 181 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745 CrossRef.
- S. Abelló, D. Verboekend, B. Bridier and J. Pérez-Ramírez, J. Catal., 2008, 259, 85 CrossRef CAS.
- B. Bridier, N. López and J. Pérez-Ramírez, J. Catal., 2010, 269, 80 CrossRef CAS.
- H. Topsøe, J. Catal., 2003, 216, 155 CrossRef CAS.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351 RSC.
- M. A. Bañares, Catal. Today, 2005, 100, 71 CrossRef CAS.
- F. C. Meunier, Chem. Soc. Rev., 2010, 39, 4602 RSC.
- E. Stavitski and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4615 RSC.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS.
- D. Teschner, Z. Revay, J. Borsodi, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Milroy, S. D. Jackson, D. Torres and P. Sautet, Angew. Chem., Int. Ed., 2008, 47, 9274 CrossRef CAS.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter, 1999, 59, 15990 CrossRef.
- E. Christoffersen, P. Stoltze and J. K. Nørskov, Surf. Sci., 2002, 505, 200 CrossRef CAS.
- A. Christensen, P. Stoltze and J. K. Nørskov, J. Phys.: Condens. Matter, 1995, 7, 1047 CrossRef CAS.
- A. Christensen, A. V. Ruban, P. Stoltze, K. W. Jacobsen, H. L. Skriver, J. K. Nørskov and F. Besenbacher, Phys. Rev. B: Condens. Matter, 1997, 56, 5822 CrossRef CAS.
- B. Bridier and J. Pérez-Ramírez, J. Am. Chem. Soc., 2010, 132, 4321 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 911 CAS.
- P. H. Bolt, F. H. P. M. Habraken and J. W. Geus, J. Solid State Chem., 1998, 135, 59 CrossRef CAS.
- M. Grioni, J. B. Goedkoop, R. Schoorl, F. M. F. de Groot, J. C. Fuggle, F. Schäfers, E. E. Koch, G. Rossi, J. M. Esteva and R. C. Karnatak, Phys. Rev. B, 1989, 39, 1541 CrossRef CAS.
- T. J. Regan, H. Ohldag, C. Stamm, F. Nolting, J. Lüning, J. Stöhr and R. L. White, Phys. Rev. B, 2001, 64, 214422 CrossRef.
- E. O. F. Zdansky, A. Nilsson and N. Martensson, Surf. Sci., 1994, 310, L583 CrossRef CAS.
- R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Laegsgaard, B. S. Clausen, J. K. Nørskov and F. Besenbacher, Nat. Mater., 2005, 4, 160 CrossRef CAS.
- B. Bridier, N. López and J. Pérez-Ramírez, Dalton Trans., 2010, 39, 8412 RSC.
- E. Boellaard, F. Th. van de Scheur, A. M. van der Kraan and J. W. Geus, Appl. Catal., A, 1998, 171, 333 CrossRef CAS.
- E. M. Silverman, R. J. Madix and P. Derlue, Surf. Sci., 1981, 109, 127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental description of preparation, catalytic tests at ambient pressure, in situ X-ray photoelectron (XPS) and X-ray absorption (XAS) spectroscopies. Powder X-ray diffraction patterns, X-ray photoelectron spectra, and alkyne hydrogenation tests at 1 bar of specific samples. See DOI: 10.1039/c1sc00069a |
| This journal is © The Royal Society of Chemistry 2011 |