Activation of elemental S, Se and Te with uranium(III): bridging U–E–U (E = S, Se) and diamond-core complexes U–(E)2–U (E = O, S, Se, Te)†
Oanh P.
Lam
,
Frank W.
Heinemann
and
Karsten
Meyer
*
Department of Chemistry and Pharmacy, Inorganic Chemistry, University Erlangen-Nuremberg, Egerlandstraße 1, D-91058, Erlangen, Germany. E-mail: karsten.meyer@chemie.uni-erlangen.de
First published on 19th May 2011
Abstract
Trivalent uranium complexes supported by tris(aryloxide) chelating ligands, [((t-BuArO)3tacn)U] and [((AdArO)3N)U], undergo activation of sulfur and selenium in their elemental forms, generating the mid-valent U(IV)/(U(IV) complexes of the type [{((t-BuArO)3tacn)U}2(μ-E)] and [{((AdArO)3N)U}2(μ-E)] (E = S, Se). Under reducing conditions, [((AdArO)3N)U] reacts with elemental sulfur, selenium and tellurium to yield the mid-valent dinuclear bis-μ-chalcogenide complexes [Na(DME)3]2[{((AdArO)3N)U}2(μ-E)2] (E = S, Se, Te) with the diamond-core structural motif and rare inorganic chalcogenide bridging ligands. For comparison, a unique high-valent U(V)/U(V) dinuclear complex [{((AdArO)3N)U}2(μ-O)2] was also synthesized. A short uranium–uranium distance in this complex with a U(μ-O)2U diamond-core may account for the unusual temperature-dependent magnetic behavior.
Introduction
Metal chalcogenide complexes formed through activation of elemental chalcogenides have long been of interest to coordination chemists.1–5Activation of elemental chalcogens has been established for transition metal complexes such as Mo,1,6,7Nb,8Ti,9Ge,10,11Zr,12 and lanthanide metal complexes.13,14 However, there are no reports on activation of elemental S, Se or Te by actinide metal complexes. The bonding involving actinide ions tends to be predominantly ionic when hard ligands, such as nitrogen and oxygen donors, are employed. Hence, studying the bonding of actinides to softer sulfide, selenide and telluride ligands could help address the question of covalency in actinide complexes.15–18 Most chalcogenide complexes of the actinides contain organochalcogenide ligands with few containing purely inorganic chalcogenide ligands.19–24 Of these, actinide complexes containing inorganic selenide and telluride ligands are the most scarce.20,21 The lack of such compounds may be attributed to the difficulties associated with activation of elemental selenium and tellurium.Recently, we have reported the synthesis and reactivity of the oxo-bridged [{((t-BuArO)3tacn)U}2(μ-O)] (2).25 In our attempts to understand the electronic structure and magnetic properties of dinuclear uranium complexes, we intended to synthesize a series of single-atom bridged dinuclear complexes, systematically varying the nature of the bridging ligand and uranium–uranium distance.
In doing so, we have discovered that trivalent uranium complexes supported by our single nitrogen and triazacyclononane-anchored tris(aryloxide) chelating ligands will react with the heavier group VI chalcogens. For instance, the activation of elemental sulfur and selenium can be achieved by employing highly reactive U(III) complexes supported by the tris-aryloxide chelating ligands [((t-BuArO)3tacn)U] and [((AdArO)3N)U] to form uranium chalcogenide-bridged complexes (Chart 1). Under reducing conditions, tetravalent dinuclear dianionic bis-μ-E (E = S, Se, Te) complexes of uranium can also be obtained. For comparison, a pentavalent uranium bis-μ-O was also synthesized. Herein, we report the syntheses, molecular structures and electronic properties of a series of uranium complexes containing purely inorganic oxide, sulfide, selenide and telluride ligands.
 | ||
| Chart 1 Chelating ligands employed for stabilization of reactive U(III) complexes, (t-BuArOH)3tacn3− (L1, left), (AdArOH)3N3− (L2, right) and the numbering scheme for U chalcogenide complexes presented herein. | ||
Results and discussion
Syntheses and molecular structures of 3 and 4
Treating the dark red-brown colored, electron-rich U(III) complex [((t-BuArO)3tacn)U] (1), ((t-BuArO)3tacn3− = trianion of 1,4,7,-tris(3,5-di-tert-butyl-2-hydroxybenzyl)-1,4,7-triazacyclononane), with 1/8 equiv. of elemental sulfur (S8) results in the formation of a bright yellow precipitate in a yellow solution (Scheme 1, top). The fine yellow solid was isolated by filtration with a yield of 58%. Recrystallization from a saturated toluene solution produced single crystals suitable for X-ray diffraction (XRD). The molecular structure reveals a dinuclear uranium complex with a bridging S2− ligand [{((t-BuArO)3tacn)U}2(μ-S)] (3) (Fig. 1, top).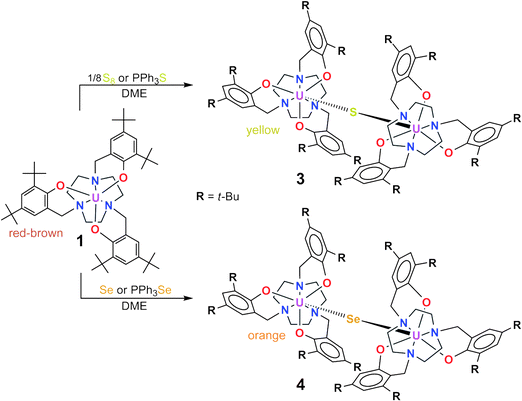 | ||
| Scheme 1 Syntheses of U(IV) sulfide-bridged (3) and U(IV) selenide-bridged (4) complexes of the tacn-based ligand system. | ||
 | ||
| Fig. 1 Molecular structures of 3 (top) and 4 (bottom). The tert-butyl groups, hydrogen atoms and co-crystallized solvent molecules were omitted for clarity. Thermal ellipsoids are at 50% probability. | ||
In contrast, the reaction of trivalent complex 1 with elemental selenium proceeds slowly, forming an orange precipitate over the course of two days. A shorter reaction time and better yield of the product can be achieved when 1 is treated with 1 equiv. of a selenide transfer reagent, namely PPh3Se, whereupon a bright orange precipitate forms within minutes (Scheme 1, bottom). The product is collected by filtration with a yield of 58%. Orange single crystals suitable for XRD analysis were obtained from diffusion of acetonitrile into a solution of 4 in benzene. The molecular structure features two U(IV) centers bridged by a Se2− ligand [{((t-BuArO)3tacn)U}2(μ-Se)] (4) (Fig. 1, bottom). All complexes, 2, 3 and 4 feature the chalcogenide ligands coordinated at axial positions of the U(IV) centers, where a C3 symmetric environment is retained at the seven-coordinate U ion. The uranium to ligand bond distances in complexes 3 (U–Navg = 2.659 Å, U–Oavg = 2.203) and 4 (U–Navg = 2.676 Å, U–Oavg = 2.206) are similar to each other and also to six-coordinate complex 1. By contrast, these distances are significantly shorter than those of the previously published uranium oxo-bridged complex [{((t-BuArO)3tacn)U}2(μ-O)]25 (2) (U–Navg = 2.746 Å, U–Oavg = 2.225 Å) (Table 1). This lengthening of the uranium to ligand distances in 2 is most likely due to an increase in uranium to ligand orbital involvement and electrostatic interaction to a much harder, stronger O2− ligand rather than to the soft S2− and Se2− ligands. Similarly, the observed increase in U–E (E = O, S, Se) distances from 2.110(4) Å (2), to 2.592(6) Å (3), to 2.7188(4) Å (4) is apparent and attributed to the increase in size and softness of the ligands as one descends the chalcogenide group. An informative parameter to examine is the uranium out-of-plane shift (Uoop), which measures the distance from the uranium center to the plane formed by the three phenolate oxygen atoms. This Uoop shift is a sensitive indicator of the strength of the U–Lax bond in these complexes: the stronger the axial seventh ligand is bound, the closer the uranium is pulled towards the plane. In moving from the O2− ligand in 2 to the S2− in 3, the uranium center shifts away from the plane by almost twice the distance (−0.078 to −0.163 Å). This observation is consistent with an increase of almost 0.5 Å in the U–O distance in 2 to the U–S distance in 3. The linearity of the U–E–U angle, also crystallographically imposed, and short U–E distances indicate that the chalcogenide group in each complex acts as a π-donor ligand.20
| Structural parameter | 2 | 3 | 4 |
|---|---|---|---|
| U–Ntacn, avg | 2.746 | 2.659 | 2.676 |
| U–OAr, avg | 2.225 | 2.203 | 2.206 |
| U–E | 2.110(4) | 2.592(6) | 2.7188(4) |
| U1⋯U2 | 4.219 | 5.183 | 5.438 |
| U–E–U | 180 | 180 | 180 |
| Uout–of–plane shift | −0.078 | −0.163 | −0.183 |
Syntheses and molecular structures of 7 and 8
In contrast to the tacn-derivatized complex 1, the U(III) complex [((AdArO)3N)U] (5), supported by the single nitrogen-anchored ligand ((AdArO)3N3− = trianion of tris(2-hydroxy-3-adamantyl-5-methylbenzyl)amine), shows remarkable reactivity towards CO2 through a reactive, sterically less shielded, oxo-bridged intermediate, [{((AdArO)3N)U}2(μ-O)] (6), to form the dinuclear carbonate complex [{((AdArO)3N)U}2(μ-η1:κ2-CO3)].26 Treating complex 5 with 1/8 equiv. of elemental sulfur (S8) results in immediate formation of a yellow–orange precipitate in an orange solution (Scheme 2, top). The isolated precipitate was characterized as the U(IV)/U(IV) μ-sulfido complex [{((AdArO)3N)U}2(μ-S)] (7). | ||
| Scheme 2 Syntheses of U(IV)/U(IV) sulfide-bridged (7) and U(IV)/U(IV) selenide-bridged (8) complexes of the single N-anchored ligand system. | ||
When complex 5 is treated with 1 equiv. of elemental selenium, a yellow–orange precipitate is formed from a red–brown suspension (Scheme 2, bottom) within 1 h, clearly indicating that 5 is much more reactive than the analogous tacn-derivatized U(III) complex 1. The yellow–orange solids were isolated and characterized as the dinuclear selenide-bridged complex [{((AdArO)3N)U}2(μ-Se)] (8). Single crystals suitable for XRD analyses of both compounds 7 and 8 were grown from saturated solutions of 1,2-dimethoxyethane (DME). Similar to 3 and 4, the molecular structures of 7 and 8 are isostructural (Fig. 2). In contrast to 3 and 4, 7 and 8 feature positive Uoop values, where the uranium centers lie above the tris-aryloxide plane formed by the three oxygens. Additionally, the S2− and Se2− ligands in 3 and 4 are coordinated at the axial position, while the S2− and Se2− ligands in 7 and 8 coordinate in the equatorial position, cis to the nitrogen anchor, rendering the coordination polyhedra of 7 and 8 as distorted monocapped trigonal prisms. The change in coordination environment from trigonal in 5 to tetragonal in 7 and 8 is indicative of the flexibility of the single nitrogen-anchored ligand in 5, a characteristic not observed in 1, where the triazacyclononane anchor maintains a rigid trigonal ligand framework. The U–N and average tris-aryloxide U–O distances of complex 7 (2.558(6), 2.185 Å) are comparable to 8 (2.556(6), 2.178 Å). Similar to the U–E–U series of the (t-BuArO)3tacn3− ligand system, an increase in U–E distances from 2.104(1) Å (6), to 2.736(2) and 2.713(2) Å (7) and to 2.830(1) and 2.816(1) Å (8) is the consequence of increasing size and softness of the chalcogenide as the group is descended (Table 2). Notably, the slightly bent U–E–U angles in 7 (173.5°) and 8 (174.2°) compared to linear 3 and 4 are reflected in the significantly longer U–E bonds observed for 7 (avg. 2.725 Å) and 8 (avg. 2.823 Å) than for 3 (2.592 Å) and 4 (2.716 Å). While the tert-butyl groups in 3 and 4 are arranged in a staggered conformation, which allows for a linear U–E–U angle, the bent U–E–U angles in 7 and 8 can be attributed to the steric congestion caused by the adamantyl substituents that prevents optimum orbital overlap in the U–E–U fragment. As expected, no trends are observed in the uranium out-of-plane shifts of 7 and 8, since the coordination sites of the chalcogenide ligands are equatorial and not axial. Recently, we have reported on the formation of a bridging carbonate complex from reductive cleavage of CO2, showing that the reaction proceeds through an oxo-bridged intermediate [{((AdArO)3N)U}2(μ-O)] (6).26 This observation is unusual since lanthanides and actinides often form thermodynamically stable bridging oxo complexes. For instance, the uranium oxo-bridged complex [{((t-BuArO)3tacn)U}2(μ-O)] (2) is completely inert. The significantly increased reactivity of 6 can be attributed to its coordinative unsaturation and the flexibility of the (AdArO)3N3− ligand. Both factors promote equatorial binding of the fifth ligand plus a weakly bound solvate molecule, DME or Et2O; thus, leaving access to the bridging oxo moiety for further chemistry. Structurally, complexes 7 and 8 are very similar to 6. Reactivity studies of these complexes are currently underway.
 | ||
| Fig. 2 Molecular structures of 7 (top) and 8 (bottom). Adamantyl groups, hydrogen atoms and co-crystallized solvent molecules were omitted for clarity. Thermal ellipsoids are at 50% probability. | ||
| Structural parameter | 6 | 7 | 8 |
|---|---|---|---|
| U–N | 2.550(4) | 2.558(6) | 2.556(6) |
| 2.565(6) | 2.551(6) | ||
| U–OAr, avg | 2.206 | 2.185, 2.180 | 2.178, 2.179 |
| U1,2–E | 2.104(1) | 2.736(2) | 2.830(1) |
| 2.104(1) | 2.713(2) | 2.816(1) | |
| U1⋯U2 | 4.207 | 5.441 | 5.639 |
| U–E–U | 179.9 | 173.5 | 174.2 |
| U1,2 out–of–plane shift | 0.530 | 0.526 | 0.533 |
| 0.530 | 0.590 | 0.587 |
Syntheses and molecular structures of 9, 10 and 11
Treating 5 with 1/8 equiv. of sulfur (S8) in the presence of sodium amalgam resulted in precipitation of yellow-green solids from a green solution. The solids were collected by filtration and characterized as the dinuclear dianionic uranium bis-μ-sulfide complex [Na(DME)3]2[{((AdArO)3N)U}2(μ-S)2] (9) (Scheme 3, left). Analogously, bright orange dianionic U(IV)/U(IV) bis-μ-selenide [Na(DME)3]2[{((AdArO)3N)U}2(μ-Se)2] (10) and bis-μ-telluride [Na(DME)3]2[{((AdArO)3N)U}2(μ-Te)2] (11) complexes can be obtained by treating 5 with 1 equiv. of elemental selenium and tellurium, respectively, in the presence of sodium amalgam (Scheme 3, center and right). | ||
| Scheme 3 Syntheses of U(IV)/U(IV) bis-μ-sulfide (9), U(IV)/U(IV) bis-μ-selenide (10), U(IV)/U(IV) bis-μ-telluride (11) dianions of the single N-anchored ligand system. | ||
Orange single crystals for XRD of 9, 10 and 11 are obtained from diffusion of 1,2-dimethoxyethane into solutions of tetrahydrofuran. The molecular structures of 9, 10 and 11 are isostructural and feature U–(E)2–U diamond-cores, in which the uranium centers are in pseudo-octahedral environments, with the two bridging S2−, Se2− and Te2− ligands, respectively, occupying positions trans and cis to the nitrogen anchor (Fig. 3). The negative charges of the U(IV)/U(IV) dianionic complexes are counter-balanced by two sodium counterions, each solvated by three molecules of DME. Interestingly, the U–(E)2–U diamond-cores are not planar, exhibiting larger folding angles as the bridging chalcogenide gets larger, from 16.7, 19.2 to 23.5° for 9, 10 and 11, respectively. This phenomenon may be attributed to a decrease in π-donor ability as the chalcogenide group is descended. The U–N distances for 9 (2.628(6), 2.633(7) Å), 10 (2.612(5) Å) and 11 (2.614(4) Å) and the average U–O distances for 9 (2.209, 2.226 Å), 10 (2.198 Å) and 11 (2.185 Å) are comparable to those of 7 and 8 (Table 3). The U–S, U–Se and U–Te bond distances in 9, 10 and 11 are in the expected range for U(IV)–S, U(IV)–Se, U(IV)–Te bonds (∼2.69–2.71 Å, 9; ∼2.82–2.87 Å, 10; ∼3.03–3.11 Å, 11). Unlike the almost symmetrical U–S bonds in the molecular structures of 9, the U–Se and U–Te bonds in 10 and 11, respectively, are slightly asymmetrical, featuring a long U–Se bond of 2.866(1) Å and a short U–Se bond of 2.819(1) in compound 10, a long U–Te bond of 3.112(1) Å and a short U–Te bond of 3.031(1) in compound 11. Given the appropriate conditions, these longer U–Se and U–Te bonds can be potentially cleaved to generate terminal U=Se and U=Te species, complexes which are currently unknown. Such uranium terminal selenide and telluride complexes could be utilized in atom transfer chemistry.
 | ||
| Fig. 3 Molecular structures of 9 (top-left), 10 (top-right) and 11 (bottom). Hydrogen atoms and two sodium counterions each solvated by three DME solvent molecules were omitted for clarity. Thermal ellipsoids are at 50% probability. | ||
| Structural parameter | 9 | 10 | 11 | 12 |
|---|---|---|---|---|
| U1,2–N1,2 | 2.628(6), 2.633(7) | 2.612 (5), 2.612(5) | 2.614(4), 2.614(4) | 2.520(5), 2.565(5) |
| U–OArO, avg. | 2.209, 2.226 | 2.198, 2.198 | 2.185, 2.185 | 2.152, 2.138 |
| U1–E1,2 | 2.690(2), 2.714(2) | 2.819(1), 2.866(1) | 3.031(1), 3.112(1) | 1.971(4), 2.174(4) |
| U2–E1,2 | 2.711(2), 2.688(2) | 2.866(1), 2.819(1) | 3.031(1), 3.112(1) | 2.057(4), 2.325(5) |
| U1⋯U2 | 3.8914(6) | 4.003(1) | 4.1890(6) | 3.4346(4) |
| U1–E1,2–U2 | 92.19(7), 92.17(7) | 89.52(3), 89.52(3) | 85.99(2), 85.99(2) | 105.9(2), 108.5(2) |
| U1,2 out–of–plane shift | 0.545, 0.567 | 0.554, 0.554 | 0.544, 0.554 | 0.514, 0.605 |
| U–E2–Ufolding angle | 16.7 | 19.2 | 23.5 | 2.2 |
Synthesis and molecular structure of 12
Mononuclear U(V) oxo complexes of the [(AdArO)3tacn3−] system are accessible viamultiple-bond metathesis of U(V) imido complexes with CO2.27In contrast, a mononuclear U(V) oxo complex of the [(AdArO)3N3−] system is as of now unknown. However, treatment of 5 with 1 equiv. of pyridine N-oxide in DME resulted in a very dark orange, almost black solution. The solution was filtered and concentrated upon which dark-orange crystals formed. The product was isolated through filtration and identified as the neutral pentavalent uranium bis-μ-oxide complex [{((AdArO)3N)U}2(μ-O)2] (12) (Scheme 4).
 | ||
| Scheme 4 Synthesis of U(V)/U(V) bis-μ-oxide (12) complex through pyridine N-oxide. | ||
Dark-orange single crystals of 12 were obtained from a concentrated solution of DME. The molecular structure of 12 features two O2− ligands bridging two pentavalent uranium centers (Fig. 4). A coordinating pyridine N-oxide molecule at one of the uranium centers renders the molecule asymmetric, so that one uranium center is in a pseudo-octahedral environment while the other uranium center is in a distorted mono-capped trigonal prismatic environment. Unlike the molecular structures of 9, 10 and 11, complex 12 exhibits a nearly planar U–(E)2–U diamond-core with a small folding angle of 2.2° (Table 3). The metal to chelating ligand average U–OArO bonds of 2.152, 2.138 and U–N bonds of 2.565(5), 2.520(5) Å are all shorter than in previous 9, 10 and 11, consistent with 12 possessing harder U(V) centers. The U–O bonds are also inequivalent in 12, two shorter U1–O2 and U2–O1 bonds of 1.971(4) and 2.057(4) Å, and two longer U1–O1 and U2–O2 bonds of 2.174(4) and 2.325(5) Å are observed. This asymmetry is more pronounced than those observed in the U–E distances of complexes 10 and 11, thus, 12 can alternatively be viewed as two interacting terminal oxo species U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The U–U distance in complex 12, measuring 3.4346(4) Å, is remarkably short and can be attributed to strong, hard–hard bonding interactions between the O2− ligands and U5+ centers. This short U–U distance may account for some unusual magnetic behavior observed for 12 (see magnetism sections below).
O. The U–U distance in complex 12, measuring 3.4346(4) Å, is remarkably short and can be attributed to strong, hard–hard bonding interactions between the O2− ligands and U5+ centers. This short U–U distance may account for some unusual magnetic behavior observed for 12 (see magnetism sections below).
 | ||
| Fig. 4 Molecular structure of high-valent U(V)/U(V) bis-μ-O complex (12). Hydrogen atoms and co-crystallazed DME solvent molecules were omitted for clarity. Thermal ellipsoids are at 50% probability. | ||
Magnetism of 2, 3, 4, 6, 7 and 8
The temperature-dependent magnetic behavior of the uranium chalcogenide series for the two different ligand systems [{((t-BuArO)3tacn)U}2(μ-E)] (E = O (2), S (3), Se (4)) (top) and [{((AdArO)3N)U}2(μ-E)] (E = O (6), S (7), Se (8)) (bottom) are shown in Fig. S1(ESI†). The low-temperature (2–5 K) magnetic moments (0.4–1.0 μB) and the room temperature (300 K) magnetic moments (3.5–4.0 μB) are consistent with values typically observed for tetravalent uranium complexes. Very little difference is observed for the triazacyclononane-anchored and the N-anchored ligand systems. However, when the magnetization data are plotted as a function of χMvs. T, a marked difference is observed between the two ligand systems. Compounds 2, 3 and 4 all exhibit a maximum at 20 K in the χMvs. T plot (Fig. 5, top), a feature indicative of antiferromagnetic exchange coupling.28,30 Such a maximum is not present in the χMvs. T plot of 6, 7 and 8. Interestingly, the U–U distances in 2, 3 and 4 are comparable to those of 6, 7 and 8, and hence, do not account for the observed exchange coupling. The exchange coupling may be attributed to the striking difference in binding site of the chalcogen ligands. In 2, 3 and 4, the chalcogen ligand is bound in the axial position compared to the equatorial position in 6, 7 and 8. Perhaps the axial binding mode of the O2−, S2− and Se2− ligands in 2, 3 and 4 promotes better orbital overlap between the two U(IV) centers and the bridging E ligand, enhancing the electronic communication between the two uranium centers. To the best of our knowledge, this is the first time antiferromagnetic exchange coupling has been observed for U(IV) complexes – previously, exchange coupling has only been observed for U(V) complexes.28,30![Magnetic susceptibility plots for [{((t-BuArO)3tacn)U}2(μ-E)] (E = O (2), S (3), Se (4)) (top) and [{((AdArO)3N)U}2(μ-E)] (E = O (6), S (7), Se (8)) (bottom) plotted as a function of χMvs. T. Data were corrected for underlying diamagnetism.](/image/article/2011/SC/c1sc00151e/c1sc00151e-f5.gif) | ||
| Fig. 5 Magnetic susceptibility plots for [{((t-BuArO)3tacn)U}2(μ-E)] (E = O (2), S (3), Se (4)) (top) and [{((AdArO)3N)U}2(μ-E)] (E = O (6), S (7), Se (8)) (bottom) plotted as a function of χMvs. T. Data were corrected for underlying diamagnetism. | ||
Andersen and co-workers have reported the room temperature magnetic moments for the bridging chalcogenide series [{(MeC5H4)3U}2(μ-E)] (E = S (2.93 μB), Se (2.85 μB), Te (3.02 μB)).20 From variable-temperature SQUID magnetization data, the authors concluded that there were no magnetic interactions between the two uranium centers.20
Besides very few reports of theoretically supported, temperature-dependent magnetization studies on molecular compounds of higher nuclearity,28–30 there is a dearth of information on magnetic exchange phenomena of uranium complexes in the literature, making it difficult to interpret their complex magnetic behavior in detail.
In order to relate structure and magnetic behavior we plan to conduct in depth investigations of 2, 3 and 4 including DFT and more detailed magnetization studies.
Magnetism of 9, 10, 11 and 12
The VT SQUID data of complexes 9, 10 and 11 show temperature-dependent magnetic moment behaviors that resemble those of complexes 6, 7 and 8. The effective magnetic moments at 2 and 300 K of 9, 10 and 11 are very similar to those of 6, 7 and 8 (Fig. 6, top). Just as in the series 2–4 and 6–8, there are no easily discernable trends identified for complexes 9–11. However, comparing the magnetic behavior of all the U(IV)/U(IV) complexes 2–4, 6–11 to U(V)/U(V) complex 12 may prove to be more informative. Due to a f1 configuration, pentavalent uranium complexes exhibit a much lower magnetic moment at 300 K (∼2.0 μB) and a magnetic moment of ∼1.3–1.5 μB at low temperatures.31 The VT SQUID magnetization measurement of 12 exhibits an expected magnetic moment of 2.15 μB at 300 K (per dimer), however, the magnetic moment of 0.52 μB at 2 K is unexpectedly low for U(V)/U(V) centers (Fig. 6, bottom). Compared to the report by Rosen et al. in 1990 on superexchange coupling between two U(V) centers in the complex [{(MeCp)3U}2(μ-1,4-N2C6H4),30 there is no maximum observed in the χMvs. T plot of 12 (see ESI†), an often definitive indication of antiferromagnetic coupling. Although, along with a considerably short U–U distance (3.4346(4) Å) in 12 and low magnetic moments at 300 and 2 K, antiferromagnetic exchange coupling between the two uranium centers may still be present. However, in order to understand these behaviors more thoroughly, detailed theoretical and magnetism studies are being conducted.![Variable-temperature magnetization data of U(iv)/U(iv) complexes [Na(DME)3]2[{((AdArO)3N)U}2(μ-E)2], E = S (9), Se (10) and Te (11) (top) and U(v)/U(v) complex [{((AdArO)3N)U}2(μ-O)2] (12) (bottom). Data have been corrected for underlying diamagnetism.](/image/article/2011/SC/c1sc00151e/c1sc00151e-f6.gif) | ||
| Fig. 6 Variable-temperature magnetization data of U(IV)/U(IV) complexes [Na(DME)3]2[{((AdArO)3N)U}2(μ-E)2], E = S (9), Se (10) and Te (11) (top) and U(V)/U(V) complex [{((AdArO)3N)U}2(μ-O)2] (12) (bottom). Data have been corrected for underlying diamagnetism. | ||
Conclusions
The activation of elemental sulfur and selenium demonstrate the powerful reducing nature of the U(III) complexes [((t-BuArO)3tacn)U] (1) and [((AdArO)3N)U] (5). The uranium complexes containing purely inorganic sulfide and selenide ligands 3, 4, 7 and 8 are the first examples of activation of elemental sulfur and selenium by uranium. Moreover, the virtually linear U–E–U angles in these complexes imply a significant degree of covalency involving the metal f-orbitals and π-donating chalcogenides. Diamond-core structures [Na(DME)3]2[{((AdArO)3N)U}2(μ-E)2], E = S (9), Se (10) and Te (11) are synthesized from 5 in the presence of Na/Hg. The Te-bridged compound 11 contains the only example of a purely inorganic U–Te bond crystallographically characterized in the literature – there are only six crystallographically characterized compounds, all containing U–Te–L units (L = Ph, PR3).17,32–34 Complexes 9, 10 and 11 possess relatively short uranium–uranium distances, which may affect their low-temperature magnetic behavior. For instance, due to a short U–U distance of 3.4346(4) Å, the pentavalent bis-μ-O complex 12 shows unusual magnetic behavior at low temperatures, with an uncharacteristically low magnetic moment of 0.52 μB 2 K.In summary, we have synthesized a series of chalcogenide-bridged dinuclear uranium complexes, which will enable us to continue investigating their electronic properties through DFT and magnetization studies.
Experimental section
General methods
All experiments were performed under dry nitrogen atmosphere, using standard Schlenk techniques or an MBraun inert-gas glovebox. Solvents were purified using a two-column solid-state purification system (Glasscontour System, Irvine, CA) and transferred to the glovebox without exposure to air.Spectroscopic methods
Magnetization data of crystalline powdered samples (20–30 mg) were recorded with a SQUID magnetometer (Quantum Design) at 10 kOe (5–300 K for 3 and 4) and (2–300 K for 7, 8, 9, 10, 11 and 12). Values of the magnetic susceptibility were corrected for the underlying diamagnetic increment (χdia = −1232.40 × 10−6 cm3 mol−1 (3), −1565.00 × 10−6 cm3 mol−1 (4), −1321.93 × 10−6 cm3 mol−1 (7), −1329.93 cm3 mol−1 (8), −1269.14 cm3 mol−1 (9), −1229.14 cm3 mol−1 (10), −1632.86 cm3 mol−1 (11), −1317.4 cm3 mol−1 (12), by using tabulated Pascal constants and the effect of the blank sample holders (gelatin capsule/straw). Samples used for magnetization measurement were recrystallized multiple times and checked for chemical composition and purity by elemental analysis (C, H and N) and 1H NMR spectroscopy. Data reproducibility was also carefully checked on independently synthesized samples.Electronic absorption spectra were recorded from 200 to 2000 nm (Shimadzu (UV-3101PC)) in the indicated solvent.
Results from elemental analysis were obtained from the Analytical Laboratories at the Friedrich-Alexander-University Erlangen-Nürnberg (Erlangen, Germany) on Euro EA 3000.
Starting materials
Precursor complexes [(THF)4UI3] and [U(N(SiMe3)2)3] were prepared as described by Clark et al.35,36Uranium turnings were purchased from Oak Ridge National Laboratory (ORNL) and activated according to literature procedures.35Sulfur (≥99.5%) was purchased from Sigma-Aldrich and used as received. Selenium pellets (∼2 mm, 99.999+%) and tellurium shots (99.999%) were obtained from Sigma-Aldrich and crushed to fine slivers before usage. Triphenylphosphine sulfide (98%) and triphenylphosphine selenide (98+%) were purchased from Acros Organics and were used as received. Anhydrous 1,2-dimethoxyethane (99.5%) was purchased from Aldrich and further dried by distilling over sodium benzophenone.Synthesis of [{((t-BuArO)3tacn)U}2(μ-S)] (3)
Elemental sulfur (3.5 mg, 0.014 mmol) was suspended in hexane (2 mL) and added to a stirring solution of [((t-BuArO)3tacn)U] (1) (110 mg, 0.108 mmol). Within 10 min, a bright yellow precipitate could be observed. The reaction was allowed to proceed for at least 12 h. The yellow precipitate was collected by filtration, washed with hexane three times (3 mL), and dried in vacuo. Yield: 60 mg (0.029 mmol, 58%). Elemental analysis (%): calc. for 3, C 59.17, H 7.59, N 4.06, S 1.55; found, C 59.00, H 7.86, N 4.28, S 1.06.Synthesis of [{((t-BuArO)3tacn)U}2(μ-Se)] (4)
A suspension of triphenylphosphine selenide (36 mg, 0.105 mmol) in hexane (∼1 mL) was added to a stirring suspension of 1 (100 mg, 0.098 mmol) in hexane (∼7 mL). Within minutes, a bright orange precipitate formed in an orange solution with concurrent formation of white triphenylphosphine. After 10 min, the orange precipitate was collected by filtration, washed with hexane three times (3 mL), and dried in vacuo. Yield: 76.5 mg (0.036 mmol, 73%). Elemental analysis (%): calc. for 4, C 57.86, H 7.43, N 3.97; found, C 57.69, H 7.42, N 3.77.Synthesis of [{((AdArO)3N)U}2(μ-S)] (7)
A suspension of elemental sulfur (2.9 mg, 0.011 mmol) in DME (∼1 mL) was added to a stirring suspension of [((AdArO)3N)U] (5) (100 mg, 0.090 mmol) in DME (∼7 mL). The reaction was allowed to proceed for 12 h. The resulting yellow–orange precipitate was filtered off, washed with cold DME three times (3 mL), dried in vacuo. Yield: 62.4 mg (0.028 mmol, 62%).Alternatively, complex 7 can be synthesized from the addition of a suspension of triphenylphosphine sulfide (27 mg, 0.092 mmol) in DME (∼1 mL) to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (∼7 mL). Over 1 h, a yellow–orange precipitate emerged out of a yellow–green solution. The yellow–orange solids were collected by filtration, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 85.7 mg (0.038 mmol, 84%). Elemental analysis (%): calc. for 7·4DME, C 60.91, H 7.43, N 1.08, S 1.23; found, C 61.20, H 7.82, N 1.20, S 0.93.
Synthesis [{((AdArO)3N)U}2(μ-Se)] (8)
Selenium pellets (7 mg, 0.089 mmol), crushed into fine slivers, were added as a suspension in DME (∼1 mL) to a stirring suspension of 5 (100 mg, 0.090 mmol). After 1 h, a yellow–orange precipitate formed in a yellow–green solution. The reaction was allowed to proceed for another 7 h. The yellow–orange precipitate was collected by filtration, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 68.3 mg (0.030 mmol, 66%).Alternatively, complex 8 can be synthesized from the addition of a suspension of triphenylphosphine selenide (31 mg, 0.091 mmol) in DME (∼1 mL) to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (∼7 mL). Over 3–4 min, a yellow–orange precipitate emerged out of a yellow–green solution. The yellow–orange solids were collected by filtration, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 88.9 mg (0.039 mmol, 86%). Elemental analysis (%): calc. for 8·4DME, C 59.83, H 7.30, N 1.06; found, C 59.62, H 7.09, N 1.27.
Synthesis of [Na(DME)3]2[{((AdArO)3N)U}2(μ-S)2] (9)
Sodium amalgam (∼3 g, 1–1.5%) was added to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (7 mL). The mixture was allowed to stir for a few minutes and elemental sulfur (2.9 mg, 0.011 mmol) was then introduced. After 5 min, the reaction solution turned dark green with some precipitate. The reaction was allowed to proceed for 20 h. The resulting yellow–green precipitate was filtered off, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 75.3 mg (0.028 mmol, 62%). Elemental analysis (%): calc. for 9, C 59.13, H 7.22, N 1.04, S 2.39; found, C 59.35, H 7.06, N 1.23, S 2.42.Synthesis of [Na(DME)3]2[{((AdArO)3N)U}2(μ-Se)2] (10)
Sodium amalgam (∼3 g, 1–1.5%) was added to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (7 mL). The mixture was allowed to stir for a few minutes and elemental selenium (7 mg, 0.089 mmol) was then introduced. After 2 h, the reaction solution turned bright orange with an orange precipitate. The reaction was allowed to proceed for another 15 h. The resulting orange precipitate was filtered off, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 76.2 mg (0.027 mmol, 61%). Elemental analysis (%): calc. for 10, C 57.13, H 6.97, N 1.01; found, C 56.48, H 6.46, N 1.08.Synthesis of [Na(DME)3]2[{((AdArO)3N)U}2(μ-Te)2] (11)
Sodium amalgam (∼3 g, 1–1.5%) was added to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (7 mL). The mixture was allowed to stir for a few minutes and elemental tellurium (12 mg, 0.094 mmol) was then introduced. The reaction mixture was allowed to stir for 11 h after which a bright orange precipitate was observed. The orange product was collected by filtration, washed with cold DME three times (3 mL), and dried in vacuo. Yield: 83.8 mg (0.029 mmol, 65%). Elemental analysis (%): calc. for 11, C 55.20, H 6.74, N 0.98; found, C 55.29, H 6.88, N 1.01.Synthesis of [{((AdArO)3N)U}2(μ-O)2] (12)
Pyridine N-oxide (13 mg, 0.137 mmol) was added to a stirring suspension of 5 (100 mg, 0.090 mmol) in DME (6 mL). Over a minute, a dark-orange solution is observed. The reaction was allowed to proceed for another 5 h. The reaction mixture was filtered and volatiles were removed to afford dark–orange solids, which were then further dried under vacuum. Yield: 90.3 mg (0.042 mmol, 93%). Elemental analysis (%): calc. for 12·DME, C 62.53, H 6.59, N 1.87; found, C 62.38, H 6.88, N 1.76.Acknowledgements
The authors acknowledge the University of Erlangen – Nürnberg, the Deutsche Forschungsgemeinschaft (DFG) through the Sonderforschungsbereich SFB 583, and ME1754/2-1 for generous financial support.Notes and references
- M. Draganjac and T. B. Rauchfuss, Angew. Chem., Int. Ed. Engl., 1985, 24, 742–757 CrossRef
.
- L. C. Roof and J. W. Kolis, Chem. Rev., 1993, 93, 1037–1080 CrossRef CAS
- J. Wachter, Angew. Chem., Int. Ed. Engl., 1989, 28, 1613–1626 CrossRef
- K. D. Karlin, Prog. Inorg. Chem., 1998, 47, 1–165 CAS
- M. C. Kuchta and G. Parkin, Coord. Chem. Rev., 1998, 176, 323–372 CrossRef CAS
- F. A. Cotton and G. Schmid, Inorg. Chem., 1997, 36, 2267–2278 CrossRef CAS
- A. R. Johnson, W. M. Davis, C. C. Cummins, S. Serron, S. P. Nolan, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 1998, 120, 2071–2085 CrossRef CAS
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020–4021 CrossRef CAS
- A. Kayal, J. Kuncheria and S. C. Lee, Chem. Commun., 2001, 2482–2483 RSC
- M. C. Kuchta and G. Parkin, J. Chem. Soc., Chem. Commun., 1994, 1351–1352 RSC
- Q. Zhang, G. Armatas and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 8665–8667 CrossRef CAS
- W. A. Howard, T. M. Trnka and G. Parkin, Organometallics, 1995, 14, 4037–4039 CrossRef CAS
- D. J. Berg, C. J. Burns, R. A. Andersen and A. Zalkin, Organometallics, 1989, 8, 1865–1870 CrossRef CAS
- A. Kornienko, J. H. Melman, G. Hall, T. J. Emge and J. G. Brennan, Inorg. Chem., 2002, 41, 121–126 CrossRef CAS
- L. Karmazin, M. Mazzanti and J. Pecaut, Chem. Commun., 2002, 654–655 RSC
- M. Mazzanti, R. Wietzke, J. Pecaut, J.-M. Latour, P. Maldivi and M. Remy, Inorg. Chem., 2002, 41, 2389–2399 CrossRef CAS
- A. J. Gaunt, B. L. Scott and M. P. Neu, Angew. Chem., Int. Ed., 2006, 45, 1638–1641 CrossRef CAS
-
L. R. Morrs, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht, 2006 Search PubMed
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS
- J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1761–1765 CrossRef CAS
- A. J. Gaunt, B. L. Scott and M. P. Neu, Inorg. Chem., 2006, 45, 7401–7407 CrossRef CAS
- D. L. Perry, A. Zalkin, H. Ruben and D. H. Templeton, Inorg. Chem., 1982, 21, 237–240 Search PubMed
- L. Ventelon, C. Lescop, T. Arliguie, M. Ephritikhine, P. C. Leverd, M. Lance and M. Nierlich, Chem. Commun., 1999, 659–660 RSC
- D. A. Wrobleski, D. T. Cromer, J. V. Ortiz, T. B. Rauchfuss, R. R. Ryan and A. P. Sattelberger, J. Am. Chem. Soc., 1986, 108, 174–175 CrossRef CAS
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242–11243 CrossRef CAS
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS
- G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS
- W. W. Lukens and M. D. Walter, Inorg. Chem., 2010, 49, 4458–4465 Search PubMed
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC
- W. J. Evans, K. A. Miller, J. W. Ziller, A. G. DiPasquale, K. J. Heroux and A. L. Rheingold, Organometallics, 2007, 26, 4287–4293 CrossRef CAS
- W. J. Evans, M. K. Takase, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2009, 28, 236–243 Search PubMed
- C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Chem. Commun., 2009, 776–778 RSC
- D. L. Clark, A. P. Sattelberger and R. A. Andersen, Inorg. Synth., 1997, 31, 307 CAS
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis/NIR data for all compounds and additional SQUID magnetization plots. CCDC reference numbers 817547–817554. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00151e |
| This journal is © The Royal Society of Chemistry 2011 |