Enhanced binding strengths of acyclic porphyrin hosts with endohedral metallofullerenes†
Bruno
Grimm
a,
Julia
Schornbaum
a,
Claudia M.
Cardona
b,
John D.
van Paauwe
c,
Peter D. W.
Boyd
*c and
Dirk M.
Guldi
*a
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058, Erlangen, Germany. E-mail: dirk.guldi@chemie.uni-erlangen.de
bLuna Innovations, Inc., 521 Bridge Street, Danville, Virginia 24541, USA
cDepartment of Chemistry, The University of Auckland, Private Bag, 921019, Auckland, New Zealand. E-mail: pdw.boyd@auckland.ac.nz
First published on 18th May 2011
Abstract
The complementary use of spectroscopy and electrochemistry shed light onto the supramolecular interactions of calixarene scaffold bearing bisporphyrins 1 and 2 as hosts with a series of fullerenes –C60, Sc3N@C80, and Lu3N@C80 – as guest molecules. Importantly, the present work shows a noticeable variation in binding strength when C60 or endohedral fullerene guests are included into the bisporphyrins hosts. These sizeable differences could be clarified by computational models of the host–guest complexes, on the one hand, and a systematic investigation of the electron transfer chemistry, on the other hand. Detailed studies document an oxidative charge transfer (i.e., electron transfer from the bisporphyrin to the fullerene) for the C60 inclusion complexes, while a reductive charge transfer (i.e., electron transfer from the fullerene to porphyrin) is operative in the endohedral metallofullerene host–guest complexes.
Introduction
Artificial photosynthetic systems for practical solar fuels production must incorporate both molecular and supramolecular assemblies to collect light energy, separate charge, and transport charge to catalytic sites where water oxidation and CO2 reduction can occur. While some progress has been made on each aspect of this complex problem, researchers have not yet developed components that are both efficient and robust, and have not yet integrated the existing functional components into a working system. The design and development of light-harvesting, photo-conversion, and catalytic modules capable of self-ordering and self-assembling into an integrated functional unit will make an efficient artificial photosynthetic system possible.1–6The three-dimensional, spherical structure of fullerenes, which are made of alternating hexagons (electron rich) and pentagons (electron deficient) with diameters starting at 7.8 Å for C60, has stimulated interest in relating their properties to conventional two-dimensional π-systems.7–12 Their extraordinary electron acceptor properties, which were predicted theoretically and confirmed experimentally, have resulted in noteworthy advances in the areas of light-induced electron transfer chemistry and solar energy conversion.13–16 It is mainly the small reorganization energy of fullerenes in electron-transfer reactions that accounts for this noteworthy breakthrough.17,18 Ultra-fast charge separation together with very slow charge recombination lead to novel electron donor–acceptor systems that can form unprecedentedly long-lived radical ion-pair states with high quantum efficiencies.19
Studies with such novel electron donor–acceptor conjugate/hybrid systems have also shed light on some basic aspects of electron transfer theory (i.e., electronic coupling elements, reorganization energies, electron tunneling, etc.). Most of this fundamental work was performed with pristine fullerenes (i.e., C60, C70, C76 and C78 for oxidative charge-shift reactions) and functionalized C60 derivatives.17,18,20,21 Structural considerations are particularly beneficial for the success of fullerenes in electron-transfer reactions.22,23 The most important feature of fullerenes in this respect is their electron delocalization, which is greatest in pristine fullerenes.3,24 However, achieving radical ion pair lifetimes in the regime of seconds requires the charges to be separated spatially by, for example, relaying them along suitably linked molecular components. In this respect, new paradigms such as deep electron traps are needed.25–27 The current work is designed to make significant contributions in this area because of the fundamental nature of the proposed investigations.
The pronounced inertness of the inner surface of fullerenes, which was first predicted on the basis of molecular orbital calculations, allows reactive species to be encapsulated.28–30 This prediction was later verified by preparing N@C60, that is, atomic nitrogen inside of C60.31–33 Here, the entrapped species has been shown to have very weak interactions with the fullerene cage. Other examples of endohedral fullerenes include La@C72, La@C74, La@C82, etc.34–36 Among these mono-metallofullerenes (MMFs), M@C82 is the most abundantly yielded species. In recent years, these and other endohedral metallofullerenes have attracted wide interest not only in physics and chemistry, but also in such interdisciplinary areas as materials and biological sciences.37 The encapsulated species is found to cover group 3 metals and most lanthanide metals, as well as their nitride clusters38–47 and carbide clusters.48–54 In contrast to, for example, N@C60, electron density distribution maps suggest significant interactions between the metal ion and the fullerene cage, although this does not necessarily imply strong covalent interactions. Moreover, synchrotron X-ray diffraction, 13C NMR and ultra-high vacuum scanning tunneling microscopy reveals that the metal atoms are not in the center of the fullerene cage but prefer a close proximity to the fullerene cage.55 In summary, the electron transfer character is unambiguously confirmed. It is notable that the electron affinities of mono-metallofullerenes are much higher than those of the corresponding fullerenes.56 A major difficulty that has hampered the use and characterization of mono-metallofullerenes is that they can only be prepared in soots that consist of many isomers including empty fullerenes.57 Consequently, only a few scattered photo-physical investigations have been reported.
In contrast to the aforementioned MMF – with an open-shell electronic structure formally described as La3+@C823− – two metal atoms might also be trapped inside the fullerene cage, yielding dimetallofullerenes. It is interesting that the Ih isomer of C80 is the most highly stabilized fullerene cage – thermodynamically and kinetically – that accommodates two La (i.e., La2@C80),58,59Ce (i.e., Ce2@C80),60,61etc. The resulting electronic structure is, for example, La26+@C806−. In fact, these have the same fullerene cage (i.e., Ih-C80) and electronic state (i.e., C806−) as Sc3N@C80 (vide infra). Nevertheless, these two endohedral metallofullerenes exhibit very different physicochemical properties. The first reduction and oxidation potentials of Ih-La2@C80 are 960 mV higher and only 10 mV lower than those of Ih-Sc3N@C80, respectively.62 The soluble and relatively air-stable di-metallofullerenes constitute one of the few violations of the isolated pentagon rule. Theoretical calculations and NMR experiments of La2@C80 have shown that the two La atoms circulate freely inside the spherical Ih-C80.63
The recently developed trimetallic nitride template method of synthesizing endohedral metallofullerenes provides access to macroscopic quantities of novel materials. The trimetallic nitride method has allowed the preparation of many members of an interesting family of compounds – the trimetallic nitride fullerenes. Their general formula is A3−nBnN@Ck (n = 0–3; A, B = group III, IV and rare-earth metals; k = 68, 78 and 80). The archetypal example is Sc3N@C80. Importantly, the yields of the trimetallic nitride fullerenes exceed those of the abundant empty-cage C84, making them the third most abundant type of fullerene structure produced under normal conditions, after C60 and C70. Although the free Sc3N molecule has not been isolated, calculations predict a pyramidal structure with an optimized Sc–N bond length of 1.957 Å and a Sc–N–Sc bond angle of 99.1°. Sc3N adopts, however, a planar structure inside C80 with Sc–N distances of 1.981, 1.967 and 2.127 Å. Sc3N@C80 has the advantage that no significant interactions are thought to exist between the trimetallic cluster guest and the fullerene host. The shortest Sc–C80 distances (2.3–2.5 Å) suggest that the scandium ions are not trapped at a specific position of the fullerene, a finding that was confirmed by density functional theory calculations, which suggest that the barrier for rotation of the Sc3N unit within C80 is rather small.38,64–66
Complementary of size and maximizing the number of points of interactions are key factors in devising stable fullerene architectures, at least in the absence of alternative motifs such as hydrogen bonding, electrostatic and metal coordination. The control over the competition between host–host, guest–host and guest–guest interactions, which is particularly evident in fullerene chemistry, where, for example, C60–C60 interactions play a major role, is important in determining the structure of supramolecular ensembles.67 Utilizating topological controlled π–π associations, a porphyrin “cyclic-dimer” and a porphyrin “jaw” have been developed.68–70 The electron-rich walls of the porphyrins and their considerable contact with incumbent C60 encouraged experiments, and strong interactions were indeed detected.71–79 In both constructs, discrete van der Waals complexes are realized with a core of two porphyrins (i.e., PdP (palladium 3-pyridiyltriphenylporphyrin) or ZnP (zinc biphenyltetrahexylporphyrin)) controlling the selective C60 incorporation. More recently, a calixarene scaffold bearing two porphyrins – bisporphyrins 1 or 2 – emerged as a supramolecular host for the efficient inclusion of C60. In this approach, the systematic variation of (i) the linkage, (ii) the type of porphyrin, and (iii) the solution properties were considered and unravel factors affecting binding of C60. Interestingly, the differences in the binding constants point to a strong dependence on different solvation energies and show that the desolvation of C60 is the major key to control the inclusion.80
In the current contribution, we wish to report on the utilization of bisporphyrins 1 or 2 as a potent means to bind a series of fullerenes – C60 (3), Sc3N@C80 (4) and Lu3N@C80 (5). Importantly, fine-tuning the fullerene-porphyrin interaction energetics leads to marked differences on binding strength, which are as large as three orders of magnitude, and, in turn, allows the selective discrimination of Sc3N@C80 (4) and Lu3N@C80 (5).
Results and discussion
In this part of our investigations, we sought to take advantage of the complexation chemistry of bisporphyrins 1 and 2 (Fig. 1) and produce host/guest inclusion complexes with a number of different fullerenes (3–5). In this respect, the pocket-like structure defined by the calixarene bisporphyrin scaffolds is ideally suited to bind fullerenes and afford intriguing host-guest structures.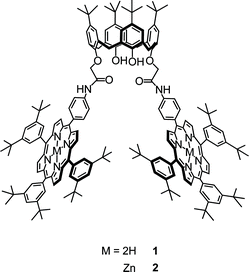 | ||
| Fig. 1 Structures of molecules used for this study. | ||
C60, Sc3N@C80 and Lu3N@C80 binding
The supramolecular inclusion of the fullerenes in the calixarene linked bisporphyrins 1 and 2 was investigated using absorption spectroscopy. To this end, constant concentrations of bisporphyrins 1 or 2 were titrated with increasing concentrations of fullerenes 3–5. By adding the endohedral fullerene 4, for example, to a solution of 1, distinct alterations in the porphyrin absorption emerge. Fig. 2 illustrates several characteristic changes as the complex 4⊂1 is formed. There is an appreciable decrease of the Soret band extinction and the emergence of two isosbestic points at 419 and 429 nm is noted. Thirdly, a minor impact on the Q-band transitions induces red-shifts. These trends are very general and both bisporphyrins give rise to similar features with C60 and the endohedral fullerenes 4 and 5. The porphyrin's π-system (1 and 2) is perturbed by electronic interactions with the different fullerenes (3–5) involving π–π interactions that are augmented by charge transfer interactions. Further novel features develop in the presence of the fullerene in the 700–900 nm range of the absorption spectra that neither originate from the two bisporphyrins nor from the different fullerenes. The following maxima were noted: 777 nm for 3⊂2 in toluene, 818 nm for 4⊂1 in ortho-dichlorobenzene, and 845 nm for 4⊂2 in ortho-dichlorobenzene. These absorptions correspond to a redistribution of charge density, namely from the electron donating bisporphyrins to the electron accepting fullerenes, leading to the formation of a charge transfer state in the 3⊂2, 4⊂1 and 4⊂2 inclusion complexes. To visualize, however, such electronic interactions mandates close electron donor–acceptor proximities. In this context, the flexibility provided by the rim of calixarene is a great asset. The latter assists in adapting intimate bisporphyrin/fullerene conformations as a means to form strongly coupled 4⊂1 (i.e., V = 20.9 cm−1 in ortho-dichlorobenzene). The energies of the charge transfer bands follow the trend seen in the electrochemically derived radical ion pair state energies – vide infra. A comparison between 3⊂1 and 3⊂2 is already published.80 | ||
| Fig. 2 Absorption titration of 1 (5.0 × 10−7 M) and various concentrations of 4 (0, 0.05, 0.10, 0.15, 0.19, 0.24, 0.33, 0.41, 0.50, 0.65, 0.83, 1.00, 1.15, 1.30, 1.43, 1.50, 1.69, 1.88, 2.01, 2.50 and 3.00 × 10−6 M) in ortho-dichlorobenzene. | ||
To correlate the spectral changes with the inclusion of the fullerenes into the cavity of the bisporphyrins Job plots were constructed from the absorption data, Fig. 3. As can be seen from a close inspection, a maximum is observed at a mole fraction value of 0.5 in the Job plot produced when 1 is allowed to interact with 3 supporting a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry for the fullerene–porphyrin complex.
:1 stoichiometry for the fullerene–porphyrin complex.
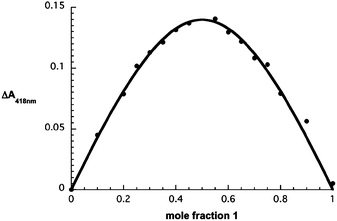 | ||
| Fig. 3 Job plot analysis in toluene–acetonitrile corresponding to the interaction of 1 and 3 with a non-linear fit function. | ||
In electrochemical experiments, bisporphyrins 1 and 2 as well as the fullerenes 3–5 revealed a number of oxidation and reduction processes and are reported vs. the Fc/Fc+ potential. In particular, oxidations at +0.24 and +0.51 V correlate well with the one-electron oxidation of 1 and 2, respectively, while reductions are seen at −1.99 and −1.65 V. The one-electron oxidation of 4 and 5 occurred at +0.5781 and +0.68 V, respectively, while that of 3 is not seen within the experimental range. One-electron reductions, on the other hand, were at −1.04 V (3) −1.25 V (4) and −1.38 (5). In decisive titration experiments in 0.1 M Bu4NClO4 containing ortho-dichlorobenzene solution, 4 and 4⊂1 were compared. Most interestingly, the presence of 1 leads to a shift of 0.29 V of the one-electron oxidation of 4. Such a shift confirms the spectroscopic absorption experiments in terms of electronically interacting 4 and 1. The magnitude of this shift indicates a significant decrease in the binding constant for 4++⊂1 in comparison to 4⊂1.82–86
Comprehensive insights into excited state interactions between the bisporphyrins (1 and 2) and the fullerenes (3–5) came from steady state fluorescence measurements. Here, the prominent fluorescence of the bisporphyrin 1 (ϕF = 0.07; τ = 9.7 ns) and 2 (ϕF = 0.05; τ = 2.0 ns) emerged as convenient tools to monitor the inclusion of the fullerene guests. Incremental addition of 3–5 leads to appreciable changes in the bisporphyrin fluorescence. The bisporphyrin fluorescence, which maximizes in the 600–750 nm range, diminishes concomitantly with the rise of a new feature centering in the 900–1100 nm range. Again, we postulate the facile formation of charge-transfer complexes, which are subject to ∼200 nm red shifts relative to the complementary absorption characteristics. These rather strong charge transfer features are discernable in non-polar as well as polar solvents with emission quantum yields of about 10−4 and emission lifetimes shorter than our time-resolution of 100 ps. For 3⊂2 the maxima shift in going from pure toluene (i.e., 963 nm) to a mixture of toluene–acetonitrile (1:1 v/v) (i.e., 1077 nm), which implies a better solvent stabilization. A somewhat surprising trend is, however, seen for 4⊂1 and 4⊂2 with maxima at 1020 and 965 nm, respectively, in ortho-dichlorobenzene. The more positive oxidation potential of 1 when compared with 2 should have resulted in an opposite trend, that is, 4⊂1 emitting in the blue and 4⊂2 emitting in the red. At this point we must conclude that a different charge transfer is operative. Importantly, the reduction potential for 1 and 4, with the latter being more negative, is compensated in 4⊂2 by the different solvent stabilization.
Fig. 4 illustrates that the quenching of the bisporphyrin fluorescence is quite drastic when 3 is titrated into a solution of 1 in toluene. Over the concentration range tested i.e. between 10−8 and 10−6 M, the fluorescence intensity drops to less than 50% of the initial value. Such changes are fully consistent with efficient excited state interactions taking place between photoexcited 1 and 3 in 3⊂1.
 | ||
| Fig. 4 Upper part – fluorescence spectra (λexc = 419 nm) of 1 (298 K, ortho-dichlorobenzene, 5.0 × 10−7 M) upon addition of different amounts of 4 (0.08, 0.15, 0.29, 0,44, 0.51, 0.71, 1.05, 1.36, 1.52, 2.01, 2.50, 3.00, 3.50, 4.00, 4.51, 5.00, 5.50 and 6.02 × 10−6 M). Lower part – plot of I/I0 for the porphyrin emission of 1 observed at 654 nm vs. concentration of 4. | ||
The observed exponential concentration/fluorescence relationship was used to quantify the association between the bisporphyrins and the fullerenes. To do this, the intensity data at 653 nm were recorded and plotted vs. the fullerene concentration.87 The binding profile obtained in this way was typical of a 1:1 association process. Nonlinear curve fitting allowed the association constant for the interaction to be calculated; the resulting Ka were in the range between 103 and 105 M−1 – see Table 1. Of particular interest is the observation that the Ka values for 1 and 2 differ by approximately two orders of magnitude for the trimetallic endohedral fullerenes (4 and 5) relative to the empty fullerene (3).
| 1 | 2 | |
|---|---|---|
| 3 | (1.87 ± 0.1) × 103 | (1.40 ± 0.1) × 103 |
| 4 | (1.34 ± 0.05) × 105 | (1.68 ± 0.1) × 105 |
| 5 | (1.57 ± 0.1) × 105 | (1.77 ± 0.1) × 105 |
The fluorescence quantum yields at the plateau values were also used to evaluate the dynamics of the excited-state deactivation process. By comparing the relative quantum yields of the bisporphyrins, for which intersystem crossing rate constants of 1.0 × 108 s−1 were calculated from the intrinsic decay of the singlet excited state, a charge-transfer rate constant of 7.0 × 109 s−1 could be calculated.
In light of the different binding constants, 3⊂1 was tested in the presence of increasing concentrations of 4 up to 2.0 × 10−5 M. We photoexcited the different samples at 432 nm corresponding to the isosbestic point in all absorption assays. In complementary fluorescence assays. With increasing concentrations of 4 the charge transfer emission of 4⊂1 grows at the expense of 3⊂1, which is only seen at lower concentrations of 4. In other words, 4 displaces 3 to form 4⊂1 – a finding that is in perfect agreement with the different Ka values seen for 3⊂1 and 4⊂1, respectively.
Computational models of host–guest complexes
Molecular models of fullerene–porphyrin host–guest complexes were computed using the ONIOM method.88,89 The calculated structures for complexes with C60 and Sc3N@C80 are shown in Fig. 5. There are several features in this host that lead to effective fullerene binding. The calix[4]arene adopts a pinched cone conformation with angles between opposing phenyl rings of 44.4 and 85.1° as is found in similar disubstituted calixarenes.90–93 In amide derivatives such as these there are two sets of hydrogen bonds between the amide N–H and the calixarene phenol oxygen (N–H⋯O) and between calixarene phenol O–H and an adjacent calixarene ether oxygen (O–H⋯O). This is observed in this model. The two planar porphyrin molecules bind to the C60 by van der Waals attraction. The porphyrins have an interplanar angle of 50.1°. The C60 is arranged with 6:6 ring junctions centered over the porphyrin at distances of 2.76, 2.67, 2.71 and 2.70 Å, as expected from arrangements in co-crystallate structures. In addition there are a significant number of C–H⋯π interactions between either the o-protons of the porphyrin mesophenyl groups or the methyl protons of t-butyl groups adjacent to the fullerene (C–H to six-membered ring centroids of C60 distances 2.7 to 3.2 Å).
 | ||
| Fig. 5 Calculated structures (ONIOM/B3LYP/6-31G(d);UFF) for the host–guest complexes of the nickel(II) derivative of 1 with (a) C60 and (b) Sc3N@C80. | ||
Similarly the two porphyrin molecules bind to Sc3N@C80. The porphyrins have a wider interplanar angle of 70.7°. The C80 is arranged with 5:6 ring junctions nearly centered over the porphyrin at distances of 2.72, 2.82, 2.92 and 2.93 Å. This centering is as found in the X-ray structure of cocrystallate of Ih-Sc3N@C80 and Co(II)octaethylporphyrin.74 As in the C60 complex there are a large number of C–H⋯π interactions from the host phenyl and tert-butyl groups to the C80 cage.
The interaction energies between a porphine molecule and the C60 or Ih-Sc3N@C80 fullerene have been estimated in the gas phase using the dispersion corrected density functional method reported by Grimme94 (C60, −19.0 kcal mol−1; Ih-Sc3N@C80, −22.2 kcal mol−1). In the case of C60 the molecule orients over the center of the porphine molecule with two carbon atoms of the 6:6 ring junction closest to the porphine plane whilst for Ih-Sc3N@C80 two carbon atoms from a 5:6 ring junction are centered over the porphine. In both cases this is found with the arrangements of these fullerenes in cocrystallates. The increased interaction energy (3.2 kcal mol−1) for Ih-Sc3N@C80 is also in qualitative agreement with the measured binding constants for the bisporphyrin hosts in solution.
Photophysical properties of host–guest complexes
Femtosecond transient absorption spectroscopy was used to obtain further insights into the excited-state interactions between the bisporphyrins (1 and 2) and the fullerenes (i.e.3–5). All of the systems were probed with 150 fs laser pulses. In the case of the bisporphyrins 1 and 2, the singlet excited state features emerge essentially immediately after the laser pulse as a result of an instantaneously deactivating excited state (i.e., second or triplet singlet excited state). The singlet–singlet transitions include characteristic absorption changes in the 500 to 810 nm range. In particular, a net decrease of the absorption around 540 nm is accompanied by new absorption bands between 660 and 730 nm. These features correlate with what would be expected for singlet–singlet transitions involving the ground state of the bisporphyrins. The singlet transient produced upon photoexcitation decays with lifetimes of 9.8 ns in 1 and 2.4 ns in 2 by intersystem crossing into the corresponding triplet manifold. The main spectral features of the latter state consist of maxima at 780 and 860 nm for 1 and 2, respectively. The triplet states relax slowly to the ground state within several tens to hundreds of μs. If, on the other hand, singlet oxygen is present, diffusion controlled deactivation of the triplet excited state gives rise to the efficient formation of singlet oxygen.In the case of the fullerenes 3–5, excitation at 420 nm gives rise to transient absorption changes that are dominated by marked singlet–singlet absorptions in the near-infrared. In this regard, 3 is a good showcase with a fingerprint absorption at 920 nm. Once generated, these features are subject to a rapid and quantitative intersystem crossing process to the energetically lower lying triplet excited state within 1.4 ns in toluene. The presence of the trimetallic clusters exerts a drastic impact on the intersystem crossing. To this end, the singlet lifetimes in 4 and 5 are as short as 48 ps. A similar trend is gathered when inspecting the corresponding triplet lifetimes with values of 48 μs95 and 109 ns for 3 and 4, respectively.
Transient absorption spectroscopy confirmed the proposed charge-transfer mechanism. Immediately after subjecting, for example 3⊂1, to a 150 fs laser pulse, the formation of the bisporphyrin singlet excited state is seen, as evidenced by their signatures. However, in contrast to what is observed for 1, a charge-separation event replaces the intrinsic deactivation, namely intersystem crossing, in the supramolecular ensemble 3⊂1, as detailed below. Fig. 6 illustrates that the singlet excited state of 1 produced following laser excitation is metastable and transforms rapidly (τ1/2 = 100 ps) into a new species. As this occurs, spectral signatures – characteristic of the one-electron oxidized form of 1 and the one-electron reduced form of 3 – are seen to grow at 600–800 and 1080 nm, respectively. Such a spectral evolution is consistent with a process of intraensemble charge transfer that yields a 3•−•−⊂1•+•+ radical-ion-pair state. Similarly, the 3•−•−⊂2•+•+ radical-ion-pair state is seen in complementary experiments with 2. Within the instrumental detection range of our femtosecond setup (3 ns), the 3•−•−⊂1•+•+ and 3•−•−⊂2•+•+ radical-ion-pair states decay.
 | ||
| Fig. 6 Differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (420 nm) of 1 (4.1 × 10−6 M) and 3 (2.0 × 10−5 M) in Ar-saturated toluene with a time delay of 50 ps at room temperature. | ||
Careful analyses of time profiles throughout the entire wavelength region revealed insights into the charge separation and recombination kinetics. In toluene solution the charge separation in 3⊂1 and 3⊂2 is fast with lifetimes of 100 ps and 85 ps, respectively (Table 2). Much more interesting is the trend on the charge recombination with lifetimes of 1458 ps for 3⊂1 and 535 ps for 3⊂2. Using a solvent mixture of acetonitrile and toluene increases the solvent polarity and is paralleled by subtle changes in the charge recombination kinetics. In fact, Fig. 7 exemplifies the acceleration of the charge recombination from 1458 to 535 ps for 3⊂1. Less drastic were changes for 3⊂2, where the acceleration amounts to about 35%, that is, from 535 to 350 ps in the absence and presence of acetonitrile, respectively.
| τ CS/ps | τ CR /ps | Solvent | |
|---|---|---|---|
| 3⊂1 | 100 | 1458 | Toluene |
| 3⊂1 | 80 | 914 |
Toluene–acetonitrile (5:1) |
| 3⊂2 | 85 | 535 | Toluene |
| 3⊂2 | 60 | 350 |
Toluene–acetonitrile (5:1) |
| 4⊂1 | 22 | 229 | oDCB |
| 4⊂2 | 51 | 779 | oDCB |
| 5⊂1 | 49 | 284 | oDCB |
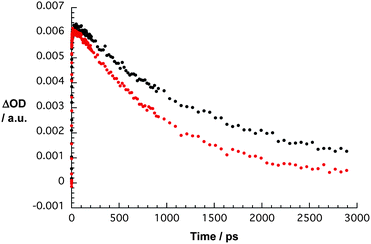 | ||
| Fig. 7 Time–absorption profile of 3⊂1 at 1080 nm in toluene (black trace) and toluene–acetonitrile (red trace), reflecting the charge separation and charge recombination. | ||
Turning to the differential absorption changes that evolve upon photoexcitation of 4⊂1 and 5⊂1, instead of seeing the spectral markers of the one-electron oxidized form of 1 and the one-electron reduced form of 4 or 5, it is the one-electron reduced form of 1 and the one-electron oxidized form of 4 or 5 that develop – see Fig. 8 and 9. To this end, the spectroelectrochemical data taken under oxidative conditions for 4 featuring maxima at 910 and 1090 nm are decisive – see Fig. S1 (ESI†). Spectroelectrochemical and pulse radiolytical evidence for the reduced 1 are the maxima at 580 and 620 nm – see Fig. S2 and S3 (ESI†). The close agreement with the photolysis experiments attests to the 4•+•+⊂1•−•− radical-ion-pair state formation.
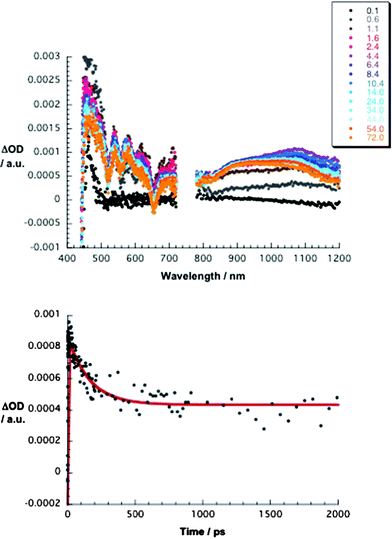 | ||
| Fig. 8 Upper part – differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (420 nm) of 1 (4.1 × 10−6 M) and 4 (2.0 × 10−5 M) in Ar-saturated ortho-dichlorobenzene with time delays between 0.1 and 72 ps at room temperature. Lower part – time–absorption profile at 1130 nm, reflecting the charge separation and charge recombination. | ||
 | ||
| Fig. 9 Upper part – differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (420 nm) of 1 (4.1 × 10−6 M) and 5 (2.0 × 10−5 M) in Ar-saturated ortho-dichlorobenzene with time delays between 0.1 and 80 ps at room temperature. Lower part – time–absorption profile at 897 nm, reflecting the charge separation and charge recombination. | ||
Conclusions
The attachment of porphyrins to the lower rim of a calix[4]arenevia an amide linker with a single methylene spacer has been shown to be most suitable for the acceptance of a fullerene guest.80 A study of the binding constants of fullerene guests into bisporphyrin hosts shows a marked variation in binding strength. Notably the weaker binding constant for [60]fullerene to the free-base bisporphyrin 1 and zinc derivative 2 was found to be about two orders of magnitude less than to the endohedral metallofullerenes. Electrochemical and excited state assays shed light onto the sizeable differences. All of the transient absorption measurements give rise to characteristic NIR absorption bands when the porphyrin–fullerene complexes are formed. In the case of C60 this is attributable to a porphyrin to fullerene charge transfer, namely 3•−•−⊂1•+•+.80 In contrast, the endohedral fullerenes show the existence of a charge transfer state with a reversal of the direction leading to the formation of 4•+•+⊂1•−•− and 5•+•+⊂1•−•− radical-ion-pair states. These acyclic host–guest complexes seem to be very suitable for contributing to the purification process of endohedral metallofullerenes. Cyclic hosts have been recently shown enhanced binding for endohedral fullerenes96,97 and to be suitable for the enrichment of higher metal-free fullerenes.98 To this end, preliminary titration experiments, in which 3⊂1 was titrated with small concentrations of 4 or 5 (i.e., ca. 1% of that of 3), reveal the efficient formation of 4⊂1 and 5⊂1, respectively at the expense of 3⊂1 as expected from the differing binding constants.Acknowledgements
Financial support by Fonds der Chemischen Industrie (FCI), Deutsche Forschungsgemeinschaft (SFB 583: Redoxaktive Metallkomplexe – Reaktivitätssteuerung durch molekulare Architekturen), and Exzellenzcluster (EAM-Engineering of Advanced Materials) is acknowledged. Work at The University of Auckland was supported by the Marsden Fund of the Royal Society of New Zealand (UOA0507) and The University of Auckland Faculty of Science Research Development Fund.Notes and references
- J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction Center, Academic Press, New York, 1993 Search PubMed.
- R. E. Blankenship, Molecular Mechanism of Photosynthesis, Blackwell Science, Oxford, 2002 Search PubMed.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051–5066 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- J. H. Weaver, Acc. Chem. Res., 1992, 25, 143–149 CrossRef CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropulos and D. R. Huffmann, Nature, 1990, 347, 354–358 CrossRef.
- J. R. Morton, F. Negri and K. F. Preston, Acc. Chem. Res., 1998, 31, 63–69 CrossRef CAS.
- M. J. Rosseinsky, J. Mater. Chem., 1995, 5, 1497–1513 RSC.
- D. M. Guldi, J. Phys. Chem. B, 2005, 109, 11432–11441 CrossRef CAS.
- H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537–546 CAS.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- N. Martin, L. Sanchez, B. Illescas and I. Perez, Chem. Rev., 1998, 98, 2527–2548 CrossRef CAS.
- F. Diederich and M. Gómez-López, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- I. Hiroshi, H. Kiyoshi, A. Tsuyoshi, A. Masanori, T. Seiji, O. Tadashi, S. Masahiro and S. Yoshiteru, Chem. Phys. Lett., 1996, 263, 545–550 CrossRef CAS.
- D. M. Guldi and K.-D. Asmus, J. Am. Chem. Soc., 1997, 119, 5744–5745 CrossRef CAS.
- H. Imahori, H. Yamada, D. M. Guldi, Y. Endo, A. Shimomura, S. Kundu, K. Yamada, T. Okada, Y. Sakata and S. Fukuzumi, Angew. Chem., Int. Ed., 2002, 41, 2344–2347 CrossRef CAS.
- D. M. Guldi and P. V. Kamat, in Chemistry, Physics and Technology, ed. K. M. Kadish and R. S. Ruoff, John Wiley and Sons, New York, 2000, pp. 225–282 Search PubMed.
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem.–Eur. J., 2003, 9, 1585–1593 CrossRef CAS.
- D. I. Schuster, Carbon, 2000, 38, 1607–1614 CrossRef CAS.
- P. J. Bracher and D. I. Schuster, in Fullerenes: From Synthesis to Optoelectronic Properties, ed. D. M. Guldi and N. Martín, Norwell, MA, 2002, pp. 163–202 Search PubMed.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- K. Tamaki, H. Imahori, Y. Sakata, Y. Nishimura and I. Yamazaki, Chem. Commun., 1999, 625–626 RSC.
- H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS.
- S. Schlundt, G. Kuzmanich, F. Spänig, G.d. M. Rojas, C. Kovacs, M. A. Garcia-Garibay, D. M. Guldi and A. Hirsch, Chem.–Eur. J., 2009, 15, 12223–12233 CrossRef CAS.
- H. Mauser, A. Hirsch, N. J. R. E. Hommes and T. Clark, J. Mol. Model., 1997, 3, 415–422 Search PubMed.
- A. Hirsch, in Topics in Current Chemistry, Springer Berlin, Heidelberg, 1999, pp. 1–65 Search PubMed.
- A. Hirsch and M. Brettreich, Fullerenes, Wiley VCH, Weinheim, 2005 Search PubMed.
- T. Almeida Murphy, T. Pawlik, A. Weidinger, M. Ĥhne, R. Alcala and J. M. Spaeth, Phys. Rev. Lett., 1996, 77, 1075 CrossRef.
- C. Knapp, K. P. Dinse, B. Pietzak, M. Waiblinger and A. Weidinger, Chem. Phys. Lett., 1997, 272, 433–437 CrossRef.
- E. Dietel, A. Hirsch, B. Pietzak, M. Waiblinger, K. Lips, A. Weidinger, A. Gruss and K.-P. Dinse, J. Am. Chem. Soc., 1999, 121, 2432–2437 CrossRef CAS.
- H. Kato, A. Taninaka, T. Sugai and H. Shinohara, J. Am. Chem. Soc., 2003, 125, 7782–7783 CrossRef CAS.
- H. Nikawa, T. Kikuchi, T. Wakahara, T. Nakahodo, T. Tsuchiya, G. M. A. Rahman, T. Akasaka, Y. Maeda, K. Yoza, E. Horn, K. Yamamoto, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 9684–9685 CrossRef CAS.
- K. Yamamoto, H. Funasaka, T. Takahashi and T. Akasaka, J. Phys. Chem., 1994, 98, 2008–2011 CrossRef CAS.
- L. Dunsch and S. Yang, Small, 2007, 3, 1298–1320 CrossRef CAS.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55–57 CrossRef CAS.
- S. Stevenson, P. W. Fowler, T. Heine, J. C. Duchamp, G. Rice, T. Glass, K. Harich, E. Hajdu, R. Bible and H. C. Dorn, Nature, 2000, 402, 427–428 Search PubMed.
- L. Dunsch, M. Krause, J. Noack and P. Georgi, J. Phys. Chem. Solids, 2004, 65, 309–315 CrossRef CAS.
- M. Krause, J. Wong and L. Dunsch, Chem.–Eur. J., 2005, 11, 706–711 CrossRef CAS.
- S. Yang and L. Dunsch, J. Phys. Chem. B, 2005, 109, 12320–12328 CrossRef CAS.
- M. N. Chaur, F. Melin, B. Elliott, A. J. Athans, K. Walker, B. C. Holloway and L. Echegoyen, J. Am. Chem. Soc., 2007, 129, 14826–14829 CrossRef CAS.
- F. Melin, M. N. Chaur, S. Engmann, B. Elliott, A. Kumbhar, A. J. Athans and L. Echegoyen, Angew. Chem., Int. Ed., 2007, 46, 9032–9035 CrossRef CAS.
- M. N. Chaur, F. Melin, J. Ashby, B. Elliott, A. Kumbhar, A. M. Rao and L. Echegoyen, Chem.–Eur. J., 2008, 14, 8213–8219 CrossRef CAS.
- J. R. Pinzón, M. E. Plonska-Brzezinska, C. M. Cardona, A. J. Athans, S. S. Gayathri, D. M. Guldi, M. A. Herranz, N. MartÌn, T. Torres and L. Echegoyen, Angew. Chem., Int. Ed., 2008, 47, 4173–4176 CrossRef CAS.
- J. R. Pinzón, C. M. Cardona, M. A. Herranz, M. E. Plonska-Brzezinska, A. Palkar, A. J. Athans, N. MartÌn, A. RodrÌguez-Fortea, J. M. Poblet, G. Bottari, T. Torres, S. S. Gayathri, D. M. Guldi and L. Echegoyen, Chem.–Eur. J., 2009, 15, 807 Search PubMed.
- C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS.
- T. Inoue, T. Tomiyama, T. Sugai, T. Okazaki, T. Suematsu, N. Fujii, H. Utsumi, K. Nojima and H. Shinohara, J. Phys. Chem. B, 2004, 108, 7573–7579 CrossRef.
- Y. Iiduka, T. Wakahara, T. Nakahodo, T. Tsuchiya, A. Sakuraba, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, T. Kato, M. T. H. Liu, N. Mizorogi, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 12500–12501 CrossRef CAS.
- Z.-Q. Shi, X. Wu, C.-R. Wang, X. Lu and H. Shinohara, Angew. Chem., Int. Ed., 2006, 45, 2107–2111 CrossRef CAS.
- Y. Iiduka, T. Wakahara, K. Nakajima, T. Tsuchiya, T. Nakahodo, Y. Maeda, T. Akasaka, N. Mizorogi and S. Nagase, Chem. Commun., 2006, 2057–2059 RSC.
- H. Yang, C. Lu, Z. Liu, H. Jin, Y. Che, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 17296–17300 CrossRef CAS.
- A. A. Popov, L. Zhang and L. Dunsch, ACS Nano, 2010, 4, 795–802 CrossRef CAS.
- S. Guha and K. Nakamoto, Coord. Chem. Rev., 2005, 249, 1111–1132 CrossRef CAS.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514–7538 CrossRef CAS.
- Z. S. Ying, C. Jin, R. L. Hettich, A. A. Puretzky, R. E. Haufler and R. N. Compton, in Fullerenes: recent advances in the chemistry and physics of fullerene and related materials, ed. K. Kadish and R. Ruof, Electrochemical Society, Pennington, NJ, 1994 Search PubMed.
- T. Suzuki, Y. Maruyama, T. Kato, K. Kikuchi, Y. Nakao, Y. Achiba, K. Kobayashi and S. Nagase, Angew. Chem., Int. Ed. Engl., 1995, 34, 1094–1096 CrossRef CAS.
- M. M. Alvarez, E. G. Gillan, K. Holczer, R. B. Kaner, K. S. Min and R. L. Whetten, J. Phys. Chem., 1991, 95, 10561–10563 CrossRef CAS.
- J. Ding and S. Yang, Angew. Chem., Int. Ed. Engl., 1996, 35, 2234–2235 CrossRef CAS.
- E. G. Gillan, C. Yeretzian, K. S. Min, M. M. Alvarez, R. L. Whetten and R. B. Kaner, J. Phys. Chem., 1992, 96, 6869–6871 CrossRef CAS.
- Y. Iiduka, O. Ikenaga, A. Sakuraba, T. Wakahara, T. Tsuchiya, Y. Maeda, T. Nakahodo, T. Akasaka, M. Kako, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 9956–9957 CrossRef CAS.
- T. Akasaka, S. Nagase, K. Kobayashi, M. Wälchli, K. Yamamoto, H. Funasaka, M. Kako, T. Hoshino and T. Erata, Angew. Chem., Int. Ed. Engl., 1997, 36, 1643–1645 CrossRef CAS.
- M. M. Olmstead, A. de Bettencourt-Dias, J. C. Duchamp, S. Stevenson, H. C. Dorn and A. L. Balch, J. Am. Chem. Soc., 2000, 122, 12220–12226 CrossRef CAS.
- K. Kobayashi, Y. Sano and S. Nagase, J. Comput. Chem., 2001, 22, 1353–1358 CrossRef CAS.
- J. M. Campanera, C. Bo, M. M. Olmstead, A. L. Balch and J. M. Poblet, J. Phys. Chem. A, 2002, 106, 12356–12364 CrossRef CAS.
- D. M. Guldi and N. Martín, J. Mater. Chem., 2002, 12, 1978–1992 RSC.
- K. Tashiro, T. Aida, J.-Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 1999, 121, 9477–9478 CrossRef CAS.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704–10705 CrossRef CAS.
- M.-S. Liao, J. D. Watts and M.-J. Huang, J. Phys. Chem. B, 2007, 111, 4374–4382 CrossRef CAS.
- Y. Sun, T. Drovetskaya, R. D. Bolskar, R. Bau, P. D. W. Boyd and C. A. Reed, J. Org. Chem., 1997, 62, 3642–3649 CrossRef CAS.
- M. M. Olmstead, D. A. Costa, K. Maitra, B. C. Noll, S. L. Phillips, P. M. Van Calcar and A. L. Balch, J. Am. Chem. Soc., 1999, 121, 7090–7097 CrossRef CAS.
- P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487–10495 CrossRef CAS.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55–57 CrossRef CAS.
- T. Ishii, N. Aizawa, M. Yamashita, H. Matsuzaka, T. Kodama, K. Kikuchi, I. Ikemoto and Y. Iwasa, J. Chem. Soc., Dalton Trans., 2000, 4407 RSC.
- P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS.
- K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189 RSC.
- M. Yanagisawa, K. Tashiro, M. Yamasaki and T. Aida, J. Am. Chem. Soc., 2007, 129, 11912–11913 CrossRef CAS.
- M. Schmittel, B. He and P. Mal, Org. Lett., 2008, 10, 2513–2516 CrossRef CAS.
- A. Hosseini, S. Taylor, G. Accorsi, N. Armaroli, C. A. Reed and P. D. W. Boyd, J. Am. Chem. Soc., 2006, 128, 15903–15913 CrossRef CAS.
- Another one-electron oxidation that refers to the D5h isomer of 4 was observed at + 0.36 V vs.Fc/Fc+. The one-electron oxidation potential of 4 at + 0.57 V vs.Fc/Fc+ corresponds to the Ih isomer.
- The ratio of the binding constants (Ka) was determined from the equation: ln(Ka/Ka+) = nF|EF − EC|/RT, where EF is the oxidation potential of the free guest and EC the oxidation potential of the complexed guest. For further information see: (a) A. E. Kaifer; M. Gómez-Kaifer. Supramolecular Electrochemistry, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) Y. Kashiwagi, H. Imahori, Y. Araki, O. Ito, K. Yamada, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 5515–5522 CrossRef CAS; (c) B. Branchi, V. Balzani, P. Ceroni, M. C. Kuchenbrandt, F. Klamer, D. Blaser and R. Boese, J. Org. Chem., 2008, 73, 5839–5851 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, John Wiley & Sons, Inc., New York, 1980 Search PubMed.
- M. H. Schmidt, G. M. Miskelly and N. S. Lewis, J. Am. Chem. Soc., 1990, 112, 3420–3426 CrossRef CAS.
- J. R. Sandifer, Electroanalytical Techniques, John Wiley & Sons, Inc., New York, 2000 Search PubMed.
- A. E. Kaifer and M. Gómez-Kaifer, Supramolecular Electrochemistry, Wiley-VCH, Weinheim, 2001 Search PubMed.
- B. Valeur, Molecular Fluorescence, Wiley-VCH, Weinheim, 2002 Search PubMed.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. MontgomeryJr., K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS.
- M. Dudic, P. Lhotak, I. Stibor, H. Petrickova and K. Lang, New J. Chem., 2004, 28, 85–90 RSC.
- I. Stibor, M. Ruzickova, R. Kratky, M. Vindys, J. Havlicek, E. Pinkhassik, P. Lhotak, A. R. Mustafina, Y. E. Morozova, E. Kazakova and V. P. Gubskaya, Collect. Czech. Chem. Commun., 2001, 66, 641–662 CrossRef CAS.
- R. Joseph, B. Ramanujam, A. Acharya, A. Khutia and C. P. Rao, J. Org. Chem., 2008, 73, 5745–5758 CrossRef CAS.
- S. K. Kim, S. H. Kim, H. J. Kim, S. H. Lee, S. W. Lee, J. Ko, R. A. Bartsch and J. S. Kim, Inorg. Chem., 2005, 44, 7866–7875 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef.
- D. K. Palit, A. V. Sapre, J. P. Mittal and C. N. R. Rao, Chem. Phys. Lett., 1992, 195, 1–6 CrossRef CAS.
- G.n. Gil-Ramírez, S. D. Karlen, A. Shundo, K. Porfyrakis, Y. Ito, G. A. D. Briggs, J. J. L. Morton and H. L. Anderson, Org. Lett., 2010, 12, 3544–3547 CrossRef CAS.
- L. P. Hernàndez-Eguía, E. C. Escudero-Adán, J. R. Pinzón, L. Echegoyen and P. Ballester, J. Org. Chem., 2011, 76, 3258–3265 Search PubMed.
- Y. Shoji, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2004, 126, 6570–6571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c0sc00569j |
| This journal is © The Royal Society of Chemistry 2011 |