A DNA-based strategy for dynamic positional enzyme immobilization inside fused silica microchannels†
TuHa
Vong
ab,
Sanne
Schoffelen
a,
Stijn F. M.
van Dongen
a,
Teris A.
van Beek
b,
Han
Zuilhof
*b and
Jan C. M.
van Hest
*a
aRadboud University Nijmegen, Department of Organic Chemistry, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: J.vanHest@science.ru.nl; Fax: +31 (0)24 3653393; Tel: +31 (0)24 3653204
bWageningen University and Research, Department of Organic Chemistry, Dreijenplein 8, 6703 HB, Wageningen, The Netherlands. E-mail: Han.Zuilhof@wur.nl; Tel: +31 (0) 317 482361
First published on 20th April 2011
Abstract
A three-enzyme cascade reaction was successfully realized in a continuous flow microreactor. The first enzyme (Candida antarctica lipase B, also known as Pseudozyma antarctica lipase B) and the third enzyme (horseradish peroxidase) of the cascade process were immobilized in a mild non-contact manner via ssDNA-ssDNA interaction in discrete zones on the capillary wall, whereas the second enzyme (glucose oxidase) was kept in the mobile phase. The unique combined feature of patterning, possibility of loading and stripping, and modularity in a fused silica microchannel is demonstrated. By changing the distance between the two enzyme patches, the reaction time available for glucose oxidase could be independently and modularly varied. The reusability of the enzymatic microfluidic system was shown by using the hybridization and dehybridization capabilities of DNA as a tool for subsequent enzyme immobilization and removal.
Introduction
Enzymes in microfluidic devices have been used for many applications including chemical analysis of proteins and nucleic acids, kinetic studies, biocatalysis and biosynthesis, which are elaborately discussed in several recent reviews.1,2,3,4,5,6 Microfluidic biocatalytic systems are particularly of interest for screening purposes since such devices allow the use of minute amounts of enzymes and can easily be adapted for high-throughput analysis. An important aspect that improves the versatility of enzyme microreactors is enzyme immobilization, for at least four reasons: enabling repeated use of enzymes, simplification of product separation, facilitation of continuous flow processes, and increasing the stability of the immobilized enzymes.7,8It would be furthermore highly beneficial if enzymes could be immobilized with a high level of spatial control. The advantage of this is that the reaction will only take place in defined areas, allowing control over the residence times of the reagent in the proximity of the immobilized enzymes. Localized immobilization of enzymes (or other biomolecules) generally involves patterning by mechanical contact, such as stamping,9 spotting,10 or grafting11 which requires direct contact with the surface. Patterning in an enclosed environment, for instance capillaries, however, requires non-contact techniques, such as electrochemical patterning12 or UV-patterning. The first method mentioned however, requires a specially designed microreactor, which has a built-in electrode to provide the necessary electrical current. UV-irradiation on the other hand, needs only a UV-transparent substrate. The benefit of using UV-transparent substrates, such as quartz or fused silica, is that they are very well compatible with current optical detection and analysis methods, which are in general the primary methods of analysis in the field of molecular biology and biochemistry. Additional advantages of using glass-like materials are their high chemical stability, pressure resistance and the ease with which they can be modified.
The majority of the reports on UV-irradiation to pattern and immobilize enzymes onto glass-like surfaces are based on destructive lithography methods.13,14 In general an entire microchannel is functionalized with a photo-sensitive linker, which then can be locally cleaved, leaving a reactive moiety to which the enzyme or protein can be immobilized. Only few examples use UV-irradiation as a constructive method for patterning.14,15
One example demonstrates the proof-of-principle of highly defined DNA patterning within microchannels using intensive UV irradiation. Amino-terminated DNA reacts during UV-irradiation with the activated photo-active linker. This methodology has to be applied with care, however, as UV-irradiation can damage DNA.14,16 Recently, we also reported a constructive lithography method, which can be used to immobilize molecules of interest, such as enzymes, to the surface in just 3 steps.17 The abovementioned examples all immobilize ss-DNA as a non-covalent linker for the attachment of biomolecules, which has the extra benefit that the immobilization process is reversible. As a result, removal and subsequent reloading of batches of enzymes can in principle be performed without having to construct a new microreactor set-up. Furthermore, modularity could be greatly enhanced when modified microchannels can be placed in series.
The success of reversible and positional immobilization depends on the specificity of the recognition site and the strength of the binding. An example of a well-known highly specific binding interaction is that of biotin with streptavidin. Unfortunately, binding is so strong (Kd = ∼ 1014 - 1015 M) that it almost resembles a covalent bond and therefore does not make the most suitable candidate for reversible binding.6
A weaker binding is based on the interaction of nickel nitrilotriacetic acid (Ni-NTA) with the hexahistidine-tag (His-Tag). The main advantage is that this moiety allows attachment of the enzyme to the surface with a known orientation, because the His-Tag can be easily built in at the N- or C- terminus via genetic engineering.18 Recycling of the surface is easy and can be done by using a highly concentrated solution of a competitive binding reagent such as ethylenediamine tetra-acetic acid (EDTA). However, the Ni-NTA His-Tag interaction is relatively weak (Kd = ∼ 106 M) and not too specific, thus a gradual loss of the enzyme is a considerable risk.4 DNA oligonucleotides are therefore an interesting alternative to immobilize enzymes. DNA has a unique molecular recognition property, which allows it to bind to its complementary strand with high specificity and affinity. The binding constant of a short (random) oligonucleotide sequence of about 21 bases long (Kd = ∼ 10−7 to 10−9 M)19 will be somewhat between that of the biotin-streptavidin and the Ni-NTA/His-Tag interactions, making a robust yet recyclable system possible. This specific binding feature is for instance applied in DNA microarray technology, which is mainly used for DNA and RNA analyses.20
In the last decade, multiple attempts have been made to convert DNA microarrays into protein arrays using this DNA-directed immobilization (DDI) strategy.19,21,22
In the work presented here, we demonstrate the reversible immobilization of enzymes involved in a three-enzyme cascade reaction via the DDI technique, using constructive photolithography in a closed fused silica microchannel (Fig. 1). Advantages over existing procedures are: (1) no direct printing/contact (i.e. can be applied inside a channel); (2) no UV light necessary during immobilization of DNA or enzyme, avoiding decomposition; (3) the active group (ssDNA) can be regenerated in contrast to e.g. epoxide groups. The non-covalent immobilization method enables a facile reuse of the capillary microreactor. The unique modular approach followed allows for the creation of well-defined patches of enzymes with respect to loading and distance to each other. Since the distance between the immobilized first (Candida antarctica lipase B) and third (horseradish peroxidase) enzyme can be varied at will, the reaction time of the second enzyme (glucose oxidase), which is dissolved in the reagent flow, can be varied independently from the other two, which is beneficial when more insight is needed in complex multistep reactions.
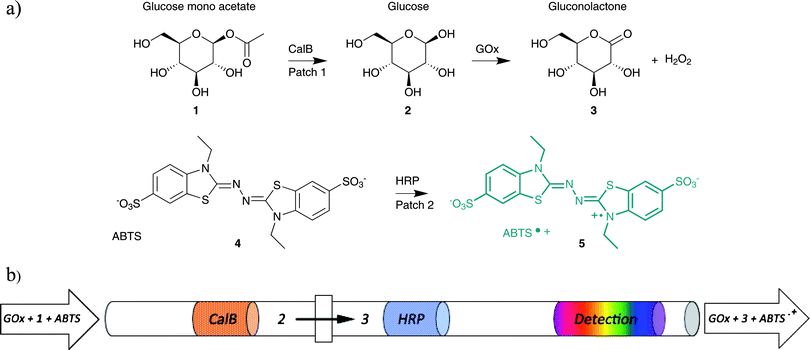 | ||
| Fig. 1 (a) Reaction scheme of the 3-enzyme cascade reaction. Glucose mono-acetate (1-O-acetyl-β-D-glucopyranose) 1 is hydrolyzed by CalB to produce glucose 2, which is subsequently oxidized by glucose oxidase (GOx) to gluconolactone 3 producing H2O2 as a side product. HRP uses H2O2 to convert ABTS (4) into ABTS•+ (5). (b) Schematic representation of the microfluidic set-up used for performing a cascade reaction. The patches represent the immobilized enzymes CalB and HRP (GOx is carried along by the mobile phase; see text) and the block represent the zero dead-volume PEEK connectors. | ||
Experimental section
General materials
Fused silica capillaries (ID = 100 μm) with a Teflon® AF fluoro-polymer external coating, TSU100375, were purchased from Polymicro (USA) and the DNA strands consisting of twenty-one bases were purchased from IBA GmbH (Germany) in lyophilized form. Candida antarctica lipase B and its azide-functionalized counterpart (AHA-CalB) as well as the azide-functionalized HRP (azido-HRP) and the substrate glucose mono-acetate (1-O-acetyl-D-gluco-pyranose, further denoted as Gluc-Ac), were obtained as described elsewhere.23,24 Horseradish peroxidase (E.C. 1.11.1.7) type VI and glucose oxidase (E.C. 1.1.3.4) type X-S from Aspergillus niger were purchased from Sigma (BioChemika). The remaining reagents and surfactants were obtained from Sigma Aldrich. Tris ((1-((O-ethyl) carboxymethyl)-(1,2,3-triazol-4-yl)) methyl) amine, further denoted as the tris-triazole ligand, used for the copper-catalyzed acetylene azide cycloaddition (CuAAC) was synthesized following literature procedures.25,26 Saline sodium citrate buffer (SSC) was obtained from VWR as a 20 × stock buffer and diluted 6.6 or 100 times to obtain 3 and 0.2 × SSC, respectively (the latter thus being 3 mM sodium citrate and 30 mM NaCl), which was used for the functionalization of the microchannel. Phosphate-buffered saline (50 mM NaH2PO4, 150 mM NaCl, calibrated with NaOH to pH 7, further denoted as PBS) was used for hybridization and kinetic studies. The DNA was purchased from IBA GmbH and used without any further purification. DNA-A: H2N-(CH2)6-5′- CCA CGG ACT ACT TCA AAA CTA-3′ (complementary strand for DNA-CalB) and DNA-B: H2N-(CH2)6-5′- GTC AAT ACT TAG GTC AAT ACT-3′(complementary strand for DNA-HRP) were used for surface attachment. DNA-AC: hexynyl-5′-TAG TTT TGA AGT AGT CCG TGG-3′ (complementary strand of DNA-A, to be used for conjugation with CalB) and DNA-BC: hexynyl-5′-AGT ATT GAC CTA AGT ATT GAC-3′(complementary strand of DNA-B, to be used for conjugation with HRP) were used to make the DNA-enzyme conjugates.Synthesis of DNA-CalB conjugate
The DNA strand used for the copper-catalyzed click reaction with the azide-functionalized CalB (AHA-CalB) was purchased with an acetylene functionality at the 5′-end and had the following sequence: hexynyl-5′-TAG TTT TGA AGT AGT CCG TGG-3′ (DNA-AC). The DNA-CalB conjugate was synthesized with DNA-AC as described.23 In short, ten eq. of acetylene-terminated DNA was added together with 15 μM of AHA-CalB (1 eq.) and the “click-mix”, consisting of 2 mM CuSO4.5 H2O (130 eq.), 2.5 mM ascorbate (170 eq.), and 4 mM tris-triazole ligand (260 eq.). This mixture was stirred overnight at 21 °C at 600 rpm. The reaction mixture was then purified by FPLC using a Superdex 75 column with PBS as eluent, and after purification used for activity determination. Protein samples were analyzed by electrophoresis on 12% (w/v) polyacrylamide gels followed by Coomassie or silver staining.Lipase activity measurements
Lipase activity of Met-CalB (natural CalB), AHA-CalB and DNA-CalB in solution was determined by the rate of hydrolysis of the substrate para-nitrophenyl butyrate (pNPB). The assays were carried out with 80 nM enzyme in PBS, 5% isopropanol and 0.1% (w/v) Triton X-100 with variable concentrations of pNPB ranging from 0.05–1.75 mM. The product formation of para-nitrophenol (pNP) was monitored via the absorbance at 405 nm over time at a set temperature of 25 °C with a Wallac Multilabel Counter 1420 UV-VIS spectrophotometer.The slope of the curve was taken as a measure of hydrolytic activity. A calibration curve of solutions with known concentrations of pNP was made to determine the specific hydrolytic activity of the different types of CalB.
Microchannel modification17
The fused silica capillaries were cleaned by consecutive flushing with acetone, ultrapure water, 1 M NaOH (1.5 h, 0.1 μL min−1) ultrapure water, 1 M HCl, ultrapure water and acetone for 20 min at 20 μL min−1 each unless stated otherwise. After drying with argon, the capillary was transferred into a glove box (MBraun MB20G, <0.1 ppm H2O, <0.1 ppm O2) under argon atmosphere and filled with neat 2,2,2-trifluoroethyl undec-10-enoate (TFEE). A mask was placed on top of the capillary leaving 6 cm of capillary uncovered. This remaining part was covered with a fused quartz microscope slide and irradiated for 10 h with two low-pressure mercury lamps (254 nm, 6.0 mW cm−2, Jelight, USA), which were placed approximately 0.5 cm above the sample. After cleaning the modified surface with petroleum ether 40°/60° and dichloromethane the capillary was filled with 50 μM of an amine functionalized DNA, which was either DNA-A (for binding with DNA-CalB) or DNA-B (for binding with DNA-HRP). The primary amino group of the ssDNA replaces the trifluoroethanol group of the activated ester in a carbonyl substitution reaction. After functionalization the unreacted DNA was removed by washing consecutively with PBST (= PBS with 0.1% Tween-20) and PBS for 20 min each at a flow of 10 μL min−1. The microchannel was now ready for the enzyme immobilisation step.The microreactor set-up for monitoring the activity of immobilized enzymes
The two microchannels with immobilized enzymes were connected with each other via a piece of unmodified fused silica tubing of variable length. Via additional pieces of fused silica tubing, the enzyme cascade was connected to a 500 μL syringe pump and the UV detector (Fig. 2). All connections were made with Upchurch Luer lock connections and zero-dead volume PEEK microtight unions (P-720). The substrate was pumped through the enzyme reactor with this syringe pump to a 45 nL flow cell (1 cm path length) in a Knauer K2501 UV-VIS detector, where detection took place. Data were further analyzed by a computer (see Fig. 2). The temperature was maintained at 25 °C with a water bath.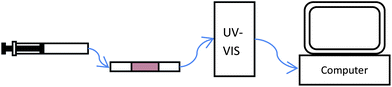 | ||
| Fig. 2 Schematic overview of the set-up used for monitoring substrate conversion of the immobilized enzyme in a capillary. | ||
Enzyme immobilization, removal and re-hybridization
A solution of DNA-CalB was flushed through the DNA- modified microchannel at RT for approximately 30 min. The microchannel was then washed with 0.2 × SSC with 0.1% sodium dodecyl sulfate (SDS). Before storage at 4 °C the solution was changed to PBS.After activity determination the DNA-CalB was removed by washing the system 30 s with 0.1 M NaOH followed by rinsing with 0.2 × SSC + 0.1% Tween-20. No activity was detected after removal. The system was regenerated by flushing with 3 × SSC for at least 2 h before re-hybridization with a fresh batch of DNA-CalB. This was done under the same conditions as the first immobilization step.
Activity determination of immobilized DNA-CalB
The activity measurements were analyzed by the hydrolysis of pNPB. The substrate mixture was kept identical to the measurements performed in bulk solution. The capillary was equilibrated with PBS and 0.1% Triton X-100 prior to use and before each measurement, the flow was set to 10 μL min−1 for 5 min before adjusting flow to the desired speed at 0.5 μL min−1. The production of pNP was monitored over time by measuring the absorbance at 405 nm, with the set-up shown in Fig. 2.To determine the converted amount of substrate, a calibration curve was made with known concentrations of pNP. The measured absorbance was recalculated to amounts of converted substrate per min per mg of enzyme. The hydrolytic activity of CalB was defined herein as μmol min−1 mg−1. N.B. More detailed information how the data was collected can be found in the electronic supplementary information†.
Preparation of DNA-HRP
The DNA-HRP conjugate was synthesized under the same conditions as the DNA-CalB. In this case the azido-HRP was coupled to DNA-BC and analyzed with SDS-PAGE. Excess of DNA and click-mix was removed by 3 spin-dialysis cycles with an Amicon® Ultra 0.5 centrifugal filter device 10,000 NMWL filter unit with MilliQ. The concentrated mixture of DNA-HRP and unreacted azido-HRP was diluted in PBS for storage. No further purification was applied. As a control, DNA-HRP and azido-HRP were rinsed through a non-functionalized capillary. Afterwards no activity could be measured when the substrate solution containing hydrogen peroxide and 2,2′-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) was applied. The same holds for the combination of azido-HRP and a DNA-modified capillary.Three-enzyme cascade reaction
The cascade reaction, as shown in Fig. 1 was performed by connecting two modified capillaries with zero-dead volume PEEK microtight unions P-720. The patches of enzymes were positioned 10 or 50 cm apart from each other. The oligo sequences used for the modified capillaries were H2N-(CH2)6-5′- CCA CGG ACT ACT TCA AAA CTA-3′ for attachment of DNA-CalB and H2N-(CH2)6-5′- GTC AAT ACT TAG GTC AAT ACT-3′ for the attachment of DNA-HRP following the procedure described above. The reaction was performed in PBS, with 2 mM Gluc-Ac, 5 mM ABTS and 100 nM GOx. Product formation (ABTS•+) was monitored by measuring the absorbance at 640 nm. Similar to the measurement with DNA-CalB, the flow was equilibrated at 10 μL min−1, before setting the desired lower flow speed.Results and discussion
Preparation of the DNA modified capillary
Fused silica capillaries were locally modified at the inside of the microchannel with ssDNA using the method previously developed by us.17,27 In comparison with our earlier work it was noticed that more DNA was attached to the surface when a higher concentration of amino-terminated DNA was used. Therefore, we increased our concentration of DNA from 5 μM to 50 μM, which was the highest workable concentration possible with the purchased amount of DNA. This improved degree of functionalization upon increasing the concentration of the nucleophilic species can be explained by the fact that the coupling of the amino-terminated DNA to the TFEE modified surface is competitive with the hydrolysis of the TFEE, due to the aqueous reaction conditions during the coupling.It was furthermore observed that within 30 min about 80% of the DNA was attached to the surface relative to a sample, which had reacted for 24 h (Fig. 3). This indicates that the coupling reaction is fast and that the first half hour of contact is critical for the amount of coupled DNA.
 | ||
| Fig. 3 Increase of the normalized fluorescence intensity over reaction time upon attachment of Cy3 labelled-DNA (relative to fluorescence obtained after 24 h of reaction (λex = 514 nm, λem = 530–600 nm). | ||
DNA-CalB conjugates
Bio-conjugation of proteins or enzymes with another moiety is often performed via attachment to cysteine and lysine residues, which contain reactive thiol and amine groups, respectively. Bi-functional cross-linkers including maleimide or N-hydroxysuccinimide groups are often preferred for their mild and effective coupling properties. However, modification via these moieties can result in changes in the structural integrity or random functionalization of enzymes, leading to possible inactive enzymes in many variations.Coupling tools that are more selective have therefore been developed, which enable targeted local introduction of unnatural amino acids.28 Examples include incorporation of alkyne and azide functionalities in proteins via genetic engineering techniques.29,30,31
Our group has used this method before to modify Candida antarctica lipase B (CalB), by replacing the methionines with azido-homoalanine residues.23 Results showed that the enzyme remained active. Moreover, it became clear that just one azido-homoalanine residue, located at the N-terminus was sufficiently exposed to be reactive towards acetylene moieties.
This knowledge was used to couple a DNA strand with an acetylene functionality (MW = 6.5 kDa) to the azido-homoalanine-modified CalB (AHA-CalB), resulting in a site-specifically modified CalB. This construct could thus be immobilized in a particular orientation at allocated areas comprising complementary DNA strands. The attachment of the DNA was analyzed with gel electrophoresis (Fig. 4a). A single band with an additional mass of ∼6.5 kDa appeared above the band of AHA-CalB after the Cu(I)-catalyzed click reaction. The product conversion was estimated to be about 70% from quantification of the signal intensity of a near-IR fluorescence scan (Fig. 4b). The reaction mixture was purified on a Fast Protein Liquid Chromatography (FPLC) system, using a size-exclusion column (Fig. 4c). The FPLC trace at 280 nm showed two peaks (see supplementary information†): one from the DNA-CalB conjugate and one from the non-reacted AHA-CalB. Together with the molecular weight increase as observed with SDS-PAGE, this indicates that indeed only one DNA strand reacted with the AHA-CalB, even though 10 equivalents were added to the reaction mixture. No free DNA was detectable in AHA-CalB after reaction and purification (Fig. 4d).
 | ||
| Fig. 4 SDS-gels displaying the MW and degree of purity of the obtained DNA-enzyme constructs, a) Coomassie stained SDS-gel of the reaction of azido-homoalanine functional CalB (AHA-CalB) with alkyne-modified ssDNA. Lane 2 AHA-CalB, lane 3 unreacted AHA-CalB (lower band) and CalB conjugated to DNA (DNA-CalB, upper band). b) Near-IR fluorescence scan of Coomassie-stained gel. Lane 2 AHA-CalB; lane 3 crude reaction mixture AHA-CalB and DNA-CalB. c) Silver-stained gel of the separate fractions after FPLC. Lane 1 CalB as reference; lane 3 DNA-CalB; lane 4 unreacted AHA-CalB. d) Agarose gel, stained with ethidium bromide, lane 1 DNA marker, lane 2 amino-terminated DNA (21 bp), lane 3 DNA-CalB after FPLC. | ||
Determination of lipase activity
The specific activity of the DNA-CalB conjugate was compared to the natural methionine-containing CalB (Met- CalB) and the engineered AHA-CalB in solution (Fig. 5). The enzyme concentration of Met-CalB or AHA-CalB was determined by measuring the absorbance at 280 nm with a Nanodrop ND-1000 spectrometer. The concentration of DNA-CalB could not be determined in this way, because DNA also absorbs in the 280 nm region, making a good estimation of the enzyme concentration with the Nanodrop impossible. Therefore, the concentration of DNA-CalB was determined by calculating the amount of converted AHA-CalB with the information obtained from the near-IR scan (Fig. 4b). | ||
| Fig. 5 Relative specific activity in solution of the CalB derivatives under current study (formation of pNP followed by absorbance at 405 nm). | ||
The conditions used for the enzyme activity assay were the same as earlier reported in order to compare results.23,32 The activities of Met-CalB, AHA-CalB and DNA-CalB were determined to be 28, 25.4 and 15.4 μmol min−1 mg−1, respectively. This relative decrease in activity of CalB after the click reaction was consistent with literature values, and was most likely caused by the reagents used during the Cu-catalyzed click reaction.33,34
It is therefore expected that more of the enzymatic activity can be retained, when Cu-free conditions, e.g. using strain-promoted cycloadditions35,36 can be used to obtain the construct, as was recently shown in an example where a PEG moiety was coupled to CalB.37
CalB activity in device
The DNA-CalB conjugate was attached to the capillary wall via hybridization with the immobilized complementary ssDNA, according to an earlier described procedure.19A fresh substrate solution was used for each measurement. The system was allowed to stabilize at 10 μL min−1 for at least 5 min before the flow was set to 0.5 μL min−1. The maximum absorbance was compared to the absorbance of solutions with known concentrations of pNP. From this calibration curve, the concentration of the converted amount of pNP was derived.
To ensure that the converted amount of pNPB is not limited by the amount of substrate, we calculated whether the diffusion distance of the substrate to the capillary wall during the residence time was not a limiting factor. This is particularly important because the enzyme is only present on the wall of the capillary. The diffusion distance of pNPB was calculated for several flow rates. For this, the diffusion constant (D) for pNPB was needed, which was obtained from the Stokes–Einstein relation (eqn (1))
 | (1) |
 | (2) |
The diffusion constant for pNPB was roughly about 2.5 × 10−10 m2 s1, i.e. typical for small molecules in an aqueous system. The diffusion distances obtainable by the residence times were calculated with the random-walk eqn (2) for the following flow rates: 0.25, 0.50 and 1.00 μL min−1 and were estimated to be 188 μm, 133 μm and 94 μm respectively (see also Supp. Info†). Since the radius of the capillary is only 50 μm, it is unlikely that the supply of pNPB to the wall-bound enzyme is limited by diffusion, as long as the flow rate is kept below 1.00 μL min−1.
With this in mind, a conservative estimation of the amount of immobilized CalB was made. To do this, the highest substrate concentration possible was used to determine the maximum amount of product formation (vmax) by the immobilized DNA-CalB. For the calculation of immobilized DNA-CalB, the assumption was made that the activity of DNA-CalB was not affected by immobilization and that the activity was the same as in bulk solution, namely 15.4 μmol pNP per min per mg DNA-CalB. Dividing these two numbers and subsequent normalization to the number of enzymes per cm2, yielded that about 2 × 1011 enzymes cm−2 were present. This corresponds to a 30% molecule coverage of the available DNA on the surface, i.e. close to what could be maximally expected given the bulkiness of the enzyme (diameter of dsDNA ∼ 2 nm; diameter CalB ∼ 5 nm).38
Reattaching a new batch of DNA-CalB conjugate
CalB is a very robust enzyme and immobilized CalB remains active even after 2 months of storage. However, the activity will decrease over time when the enzyme is continuously used. To postpone the disposal of the capillary after deactivation of the enzyme, the enzyme can be removed by dehybridization of the DNA and reattachment of new enzymes by flushing with another batch of DNA-linked enzyme. This should prolong the lifetime of the modified capillary considerably.The immobilized enzyme was removed by rinsing the capillary for 30 s with 0.1 M NaOH, after which no residual enzymatic activity could be detected. Because it is likely that the surface will be damaged after prolonged exposure to a significant concentration of NaOH, the contact time was kept as short as possible. After regeneration, rehybridization was performed under conditions identical to those of the initial hybridization step.
These stripping and reloading experiments were repeated for five cycles and performed in triplicate. After each dehybridization step, it was ascertained that the activity was zero, showing that dehybridization was complete. The normalized results, compared to the initially observed activity after the first hybridization step of this recycling method, are depicted in Fig. 6. It can be concluded from this figure that recycling is possible and about 60% of the initial activity is still maintained after five cycles, similar to literature reports on stripping and re-using DNA microarrays.39,40,41 The observed diminishing activity, however, did not lead to any mismatched hybridization.
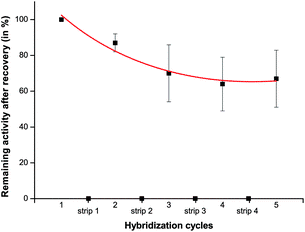 | ||
| Fig. 6 Normalized activity of the immobilized enzyme after one to five hybridization/dehybridization cycles (with respect to the first measurement). Each point represents an average of 3 measurements obtained from different microchannels with immobilized DNA-CalB. The data points strip 1–4 correspond to the activity of the microchannel after removal of DNA-CalB. | ||
Three-enzyme cascade reaction
The versatility and usability of the abovementioned method, which allows immobilization of any desired DNA-enzyme conjugate selectively at allocated areas, was subsequently demonstrated by execution of a well-studied biocatalytic cascade reaction,42 as shown in Fig. 1.For this purpose DNA-CalB was immobilized onto the first patch. GOx was left in solution as it is the “slowest” enzyme in sequence. HRP was prepared following literature procedures24 and coupled with CuAAC to hexynyl-5′-AGT ATT GAC CTA AGT ATT GAC-3′, under similar conditions as applied for DNA-CalB to obtain DNA-HRP. As previous findings showed that unreacted CalB did not bind to the modified capillaries, the DNA-HRP was only filter dialyzed to remove the excess of unreacted DNA. As can be seen in the SDS-gel in Fig. 7a, the DNA-HRP complex was formed, although not as effective as the DNA-CalB. The lower coupling efficiency is most likely due to the fact that the available azide groups of azido-HRP are sterically more hindered than the specially engineered AHA-CalB. Furthermore, the agarose gel in Fig. 7b shows that the fraction after filter dialysis did not contain any unbound DNA fragments.
 | ||
| Fig. 7 On the left a Coomassie stained SDS-gel with DNA-HRP and HRP-N3 (lane 1), HRP-N3 starting material (lane 2) and the marker (lane 3). On the right an ethidium bromide stained agarose gel. In lane 1 the DNA-HRP conjugate after 3 consecutive dialysis cycles is shown, lane 2 and 3 are non-functionalized DNA in PBS and SSC buffer, respectively, and in lane 4 a DNA marker for small fragments is visualized. | ||
The capillaries were pre-treated with 3 × SSC for 30 min before adding the DNA-CalB or DNA-HRP. The flow was set to 0.5 μL min−1 and left to bind for at least another 30 min. After binding the capillary was rinsed with PBS and PBS + 0.1% Tween-20 for another 10 min each. The capillaries were then connected in series with zero-dead volume connectors and Luer lock fittings to the syringe pump until any air bubbles had disappeared. Then the loaded capillaries were attached to the nanoflow cell of the UV-detector. As controls, (1) DNA-CalB and DNA-HRP were flushed through a non-modified capillary, (2) DNA-CalB was flushed through the capillary containing DNA-B (the non-complementary strand) and (3) this procedure was also performed for DNA-HRP through a capillary containing DNA-A. In all controls no conversion of any substrates (pNPB to pNP for DNA-CalB and ABTS + H2O2 to ABTS+• + H2O for DNA-HRP) was detected. Each experiment was started by flushing the system with PBS without any substrate, until the baseline was stable. Then the solvent was switched to the substrate mixture at a flow of 10 μL min−1 until again the baseline was stable. After this, the flow was decreased to the desired flow rate (0.25, 0.50 or 1.0 μL min−1, Fig. 8).
 | ||
| Fig. 8 Product absorbance measurement of a three-enzyme cascade reaction (CalB, GOx and HRP, where the first and third enzyme are immobilized and the second resides in the mobile phase) at different flow rates and different distances between immobilized enzyme 1 (CalB) and 2 (HRP). The patches of immobilized enzymes were 6 cm long, i.d. = 100 μm in PBS pH 7. | ||
Product formation was only detected when the enzymes were put in the correct order, whereas no product was formed when the order of CalB and HRP was reversed.
Two parameters were varied: 1) the distance – from 10 to 50 cm – between the enzyme patches (each of 6 cm width), in order to change the available reaction time for GOx independently from the immobilized enzymes; 2) the total reaction time of the cascade system, in order to conclude whether GOx is indeed the only limiting factor of the cascade reaction.
As expected, product formation was increased as the spatial distance between the first and last enzyme was enlarged. This confirms that GOx has a lower turnover speed than CalB and HRP. However, we found that a five times larger spacing did not result in five times more product, which would indicate that GOx at this prolonged reaction time is not the limiting factor anymore in the performed reaction. Moreover, the increase in conversion by enlarging the spatial distance was different for 0.25 μL min−1 than for 1.0 μL min−1. It was found that this effect was relatively larger for lower flow rates than for higher flow rates. It seems that halving the flow rate, i.e. doubling the reaction time, results in only ∼ √2 increase of end-product formation, which was observed for both spatial distances. This suggests that diffusion of at least one of the substrates could play a fairly important role in this given example. The importance of diffusion is supported by the fact that the random-walk equation shows that the diffusion distance is correlated with the diffusion time with a factor of √2 (see eqn (2)).
Multi-enzyme cascade reactions are kinetically complex. The understanding thereof thus relies on detailed knowledge of each enzyme reaction step and the influence that the previous step has on the following reaction. Other aspects that increase the complexity of a multi-enzyme cascade reaction are initial substrate concentrations, rate of substrate supply, substrate depletion and enzyme concentration.43 Therefore, there is a need for a method to vary each of the parameters independently, which can be used to increase the understanding of the system and to optimize the reaction sequence. The set-up as depicted in Fig. 1 allows the creation of well-defined patches of enzymes inside the microchannel. As a result, the amounts of enzyme present and the reaction time for a specific enzyme are controlled. Secondly, the modularity of the system allows the execution of multi-step enzymatic reactions by placing the well-defined modified capillaries in sequential order. Because the system is modular, the reaction conditions can be tailored such that reaction conditions can be adjusted independently for a single enzyme. Our findings with the three-enzyme cascade reaction show that our system can be used to obtain more insight in the kinetics of a complicated reaction sequence, which is here demonstrated by independently adjusting the reaction time of one enzyme without changing enzyme or substrate concentrations.
Conclusions
Oligonucleotide-enzyme constructs, such as DNA-CalB and DNA-HRP, both obtained by Cu-catalyzed alkyne-azide cycloadditions, were locally immobilized within discrete zones inside a microchannel, using constructive photolithographic techniques. A three-step enzymatic cascade was performed using the assembled enzymatic microreactor, placing DNA-CalB and DNA-HRP in sequential order and leaving the enzyme glucose oxidase in solution. This modular approach allows tight control of the reaction conditions and enables easy fine-tuning to get a better understanding of the role of a single enzyme within a multi-enzyme reaction.Advantages over existing procedures are that the active moieties can be applied on the inside of capillary, the enzyme immobilisation step is very mild and the enzyme, once denaturated, can be easily replaced by fresh enzyme without having to change the capillary or active moiety (ssDNA). The latter feature allows an easy and effective reusability of the microfluidic system. This was demonstrated by repetitive stripping and reloading of the microchannel.
Acknowledgements
The authors thank the General Instruments department from the Radboud University and E.J.C. van der Klift from Wageningen University and Research for the use of their equipment. NWO-ACTS (PoaC project 053.65.002), funded by the Dutch Ministry of Economic Affairs, is acknowledged for financial support.Notes and references
- P. L. Urban, D. M. Goodall and N. C. Bruce, Biotechnol. Adv., 2006, 24, 42 CrossRef CAS.
- M. Miyazaki and H. Maeda, Trends Biotechnol., 2006, 24, 463 CrossRef CAS.
- M. Miyazaki, T. Honda, H. Yamaguchi, M. P. P. Briones and H. Maeda, Biotechnol. Genet. Eng., 2008, 25, 405 Search PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775 CrossRef CAS.
- J. Krenkova and F. Svec, J. Sep. Sci., 2009, 32, 706 CAS.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453 RSC.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289 CrossRef CAS.
- T. Wilhelm and G. Wittstock, Langmuir, 2002, 18, 9485 CrossRef CAS.
- H. Schröder, L. Hoffmann, J. Müller, P. Alhorn, M. Fleger, A. Neyer and C. M. Niemeyer, Small, 2009, 5, 1547 CrossRef.
- F. Bano, L. Fruk, B. Sanavio, M. Glettenberg, L. Casalis, C. M. Niemeyer and G. Scoles, Nano Lett., 2009, 9, 2614 CrossRef CAS.
- H. Kaji, M. Hashimoto and M. Nishizawa, Anal. Chem., 2006, 78, 5469 CrossRef CAS.
- T. C. Logan, D. S. Clark, T. B. Stachowiak, F. Svec and J. M. J. Fréchet, Anal. Chem., 2007, 79, 6592 CrossRef CAS.
- B. Renberg, K. Sato, K. Mawatari, N. Idota, T. Tsukaharad and T. Kitamori, Lab Chip, 2009, 9, 1517 RSC.
- A. Arora, G. Simone, G. B. Salieb-Beugelaar, J. T. Kim and A. Manz, Anal. Chem., 2010, 82, 4830 CrossRef CAS.
- R. P. Sinha and D. P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225 RSC.
- T. H. Vong, J. Ter Maat, T. A. van Beek, B. van Lagen, M. Giesbers, J. C. M. van Hest and H. Zuilhof, Langmuir, 2009, 25, 13952 CrossRef CAS.
- Patented by Hoffmann-La Roche, EP0282042, exclusively licenced to Qiagen.
- P. Wilkins Stevens, M. R. Henry and D. M. Kelso, Nucleic Acids Res., 1999, 27, 1719 CrossRef CAS.
- C. Debouck and P. N. Goodfellow, Nat. Genet., 1999, 21, 48 CrossRef CAS.
- S. Weng, K. Gu, P. W. Hammond, P. Lohse, C. Rise, R. W. Wagner, M. C. Wright and R. Kuimelis, Proteomics, 2002, 2, 48 CrossRef CAS.
- C. M. Niemeyer, Trends Biotechnol., 2002, 20, 395 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. van Eldijk and J. C. M. van Hest, Bioconjugate Chem., 2008, 19, 1127 CrossRef CAS.
- S. F. M. van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. van Berkel, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2009, 20, 20 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS.
- S. I. van Kasteren, H. B. Kramer, D. P. Gamblin and B. G. Davis, Nat. Protoc., 2007, 2, 3185 Search PubMed.
- J. ter Maat, R. Regeling, M. Yang, M. N. Mullings, S. F. Bent and H. Zuilhof, Langmuir, 2009, 25, 11592 CrossRef CAS.
- L. Wang and P. G. Schultz, Chem. Comm., 2002, 1 RSC.
- A. J. Link and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 11164 CrossRef CAS.
- R. M. Hofmann and T. W. Muir, Curr. Opin. Biotechnol., 2002, 13, 297 CrossRef CAS.
- R. S. Goody, K. Alexandrov and M. Engelhard, ChemBioChem, 2002, 3, 399 CrossRef CAS.
- R. L. M. Teeuwen, S. S. van Berkel, T. H. H. van Dulmen, S. Schoffelen, S. A. Meeuwissen, H. Zuilhof, F. A. de Wolf and J. C. M. van Hest, Chem. Commun., 2009, 4022 RSC.
- S. C. Fry, Biochem. J., 1998, 332, 507 CAS.
- E. Lallana, E. Fernandez-Megia and R. Riguera, J. Am. Chem. Soc., 2009, 131, 5748 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97 RSC.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, Worth publishers NY, 2000, 3rd ed.; Diameter of CalB estimated from PDB file 1TCA, with N-terminus orientated downwards Search PubMed.
- Z. Hu, M. Troester and C. M. Perou, BioTechniques, 2005, 38, 121 CrossRef CAS.
- K. Hahnke, M. Jacobsen, A. Gruetzkau, J. R. Gruen, M. Koch, M. Emoto, T. F. Meyer, A. Walduck, S. H. E. Kaufmann and H. J. Mollenkopf, J. Biotechnol., 2007, 128, 1 CrossRef CAS.
- H. Wu, J. A. Bynum, S. Stavchansky and P. D. Bowman, BioTechniques, 2008, 45, 573 CrossRef CAS.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem.–Eur. J., 2009, 15, 1107 CAS.
- H. C. Hemker and P. W. Hemker, Proc. R. Soc. London, Ser. B, 1969, 173, 411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the calculations concerning the amount of immobilized enzyme and diffusion of the substrate are given there. See DOI: 10.1039/c1sc00146a |
| This journal is © The Royal Society of Chemistry 2011 |