Exploring nucleation of H2S hydrates†
Shuai
Liang
and
Peter G.
Kusalik
*
Department of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta, Canada. E-mail: peter.kusalik@ucalgary.ca
First published on 20th April 2011
Abstract
Crystal nucleation is a rare event process, typically characterized by long induction times. The direct observation of nucleation of a gas hydrate in molecular simulations has been rather challenging, where recent work has focused primarily on CH4 hydrate systems. Here we show that a H2S hydrate can nucleate very rapidly within two-phase H2O/H2S systems under relatively modest (i.e., 15–20% undercooling) thermodynamic driving forces. In agreement with observations for CH4 hydrates at high supersaturations, the homogenous nucleation process features the initial formation of amorphous hydrate-like structures, which then slowly anneal into more crystalline forms. The present results demonstrate that increased gas concentration significantly increases the probability of gas molecules exhibiting a high degree of second-neighbor coordination, thereby dramatically reducing the (average) induction time for nucleation. We confirm that the composition of aqueous solution is critical in determining its susceptibility to nucleation.
1. Introduction
Gas hydrates are crystalline compounds in which gas molecules are trapped within hydrogen-bonded water cages.1 The crystal structures of most gas hydrates can be classified into three types, two cubic structures known as sI and sII, and one hexagonal structure known as sH.1 Recently molecular simulation2 and experimental3 studies have identified another hexagonal crystalline hydrate structure called HS-I which was previously known as a hypothetical hydrate structure.4 Gas hydrates have received intense interests in the scientific and industrial fields because of their potential relevance in hydrocarbon extraction,5 energy storage,6,7CO2 sequestration,8 global warming,9,10 marine geohazards,11etc. In particular, the understanding of gas hydrate formation is crucial for energy recovery and storage, and the flow assurance in oil and natural gas pipelines. The experimental characterization of the microscopic process involved in hydrate nucleation is difficult because of the temporal and spatial limitations of current real-time monitoring techniques.12–16 Molecular dynamics (MD) simulation has become a powerful and trusted technique in providing molecular level details of phase transition processes.To this point CH4 hydrates have been the focus of most simulation studies of hydrate nucleation. MD simulations of nucleation of CH4 hydrates have been reported by several groups.17–21 Rodger and co-workers17,18 have studied a series of MD simulations of CH4 hydrate nucleation within a two-phase (an aqueous phase in contact with a CH4 gas phase) system prepared by melting a hydrate crystal. They observed that the induction time for the hydrate nucleation is related to the length of time used for melting the crystal and the resulting concentration of CH4 in the aqueous phase.18 Walsh et al.20 studied the nucleation of CH4 hydrate within a fully demixed system (i.e., separate CH4 and aqueous phases). In their work, the initial configuration was prepared by melting a CH4 hydrate, initially resulting in a two-phase (aqueous, CH4 gas) system, which was then cooled and pressurized to hydrate-forming conditions (250 K and 50 MPa), generating a spherical CH4 bubble. Even with a large effective CH4 pressure inside the nano-bubble, the observed induction time for the hydrate nucleation was still in the microsecond range.20 Vatamanu and Kusalik21 have shown that elevated local concentrations of CH4 can dramatically enhance the apparent nucleation rate of CH4 hydrates. They demonstrated that areas locally richer in CH4 will nucleate much more readily, suggesting the important role of the local gas composition in determining the nucleation behavior of the gas hydrates. Using a computationally efficient coarse-grained model and with temperatures of about 20–30% undercooling, Molinero and co-workers22,23 have successfully performed multiple independent simulations of nucleation of hydrates of guests with varied sizes and solubilities, including a methane-like guest. They found that the overall microscopic mechanism, which involves initial formation of an amorphous solid similar to that characterized in ref. 21, is similar for all guests that have been investigated.
Of all the common hydrate guests, H2S is known to form hydrates at the lowest temperature and persists to the highest temperature.24 We have recently performed MD simulations of the heterogeneous crystal growth of H2S hydrates from both two-phase (hydrate crystal, H2S aqueous solution) and three-phase (hydrate crystal, H2S aqueous solution and liquid H2S) systems.25 The H2S hydrates grown in these simulations exhibit a low level of defects, which is consistent with the relatively high stability observed experimentally for this crystal.24 We observed that the H2S hydrates can be grown at a higher rate than CH4 hydrates under comparable thermodynamic conditions.
In this report, we present observations of very rapid nucleation of H2S hydrates within two-phase H2O/H2S systems. Three systems, with a spherical H2S nano-bubble, a cylindrical H2S nano-bubble and a liquid H2S slab, are studied and all exhibit similar nucleation behavior, where nucleation is most rapid in the first case. The nucleation of the H2O/H2S systems features the rapid formation of amorphous hydrate-like structures, which then transform slowly into more recognizable crystalline forms, consistent with the two-step nucleation mechanism observed for CH4 hydrates,21 as well as coarse-grained model hydrates with guests of different sizes and solubilities.22,23 The observation of the rapid nucleation of gas hydrate from equilibrated two-phase systems under relatively modest conditions (i.e., 15% undercooling) provides clear evidence that the local composition of the aqueous solution is a critical order parameter in the nucleation of gas hydrates with small guest molecules.
2. Methods
Three systems, system A (sys-A) with 240 H2S and 1403 H2O molecules, and system B (sys-B) and system C (sys-C) with 409 H2S and 1234 H2O molecules have been studied. The sizes of sys-A, sys-B and sys-C are roughly 38 × 38 × 39 Å3, 38 × 38 × 43 Å3, and 24 × 24 × 105 Å3, respectively. In sys-A and sys-B the H2S molecules form (imperfect) spherical and cylindrical nano-bubbles within the aqueous phase, while sys-C features a liquid H2S slab. Although surface tension will contribute a significant additional pressure inside the nano-bubbles, H2S is always in the liquid phase under the present conditions. The average H2S concentrations in the aqueous solution of the three systems are presented in Table 1. We can see that the H2S concentration in sys-A is significantly higher than that in sys-B and sys-C.| X H2S | |||
|---|---|---|---|
| T/K | Sys-A | Sys-B | Sys-C |
| 260 | 0.073 (±0.008) | 0.041 (±0.005) | 0.038 (±0.004) |
| 250 | 0.077 (±0.011) | 0.044 (±0.005) | 0.040 (±0.006) |
The starting systems were equilibrated (for 10 ns) at 350 K and 50 MPa. The five statistically independent starting configurations of sys-A were then prepared at 300 K. These configurations were subsequently cooled to temperatures ranging from 210 to 255 K. All MD simulations reported here were performed with fully periodic boundary conditions, under NPT conditions at a pressure of 50 MPa. One additional simulation of sys-A was performed at 0.5 MPa (see ESI†). We note that the ice-point for the TIP4P/2005 model used here is about 248 K at 50 MPa,26 and our best estimate of the melting point, Tm, of the present model H2S hydrate system is about 290 K under the present conditions.25 Below we will refer to the undercooling, ΔT = T − Tm, where T is the simulation temperature. The simulation runs each ranging from tens to hundreds of ns were performed, depending on the times needed for a nucleation to occur. Additional details of the models and simulation methodologies used in this work can be found in ESI†.
All the molecular configurations presented in this report are averaged configurations27 over 20 ps trajectory segments, with the H2O molecules labeled as solid-like and liquid-like according to their translational mean-square displacements with respect to their averaged position.27–30 The F4 structural order parameter for each H2O molecule was averaged over all neighboring H2O molecules within 3.5 Å of the central molecule.31 To estimate the composition of the solution, a H2S cluster analysis was used to exclude H2S molecules that were part of the liquid bubble (i.e., the largest cluster). In this analysis H2S molecules with an intermolecular distance less than 4.5 Å were defined to be in the same cluster. The largest cluster in the system was always the H2S nano-bubble.
3. Results and discussion
Our detailed analyses will focus on sys-A, although the three systems show qualitatively similar behavior. Fig. 1 shows results from a typical trajectory for sys-A at ΔT = −40 K, where four distinct measures are employed to track the nucleation process within the system. These include: (A) an energetic measure based on the total potential energy of the system, (B) the structural order parameter F4 for detecting hydrate-like arrangements of H2O molecules,31 (C) the number of solid-like H2O molecules as determined by a dynamic measure based on the mean-square displacements over short trajectory segments,27–30 and (D) the H2S composition of the aqueous solution, XH2S. These various measures consistently show a transition of the system from liquid-like to solid-like behavior at around 60 ns. Molecular configurations taken from various stages of the simulation are also presented in Fig. 1(I–IV). At 30 ns we see a typical liquid configuration, at 57 ns the initial formation of some solid-like structure is apparent, while the system is clearly in its rapid growth phase at 70 ns. The final system configuration from this trajectory is shown in Fig. 1(IV), where the solid-like character of the system is evident. | ||
| Fig. 1 The evolution of four order parameters and molecular configurations from an initial two-phase H2S/H2O system (sys-A) at ΔT = −40 K. (A) The negative potential energy of the system. (B) The F4 structural order parameter of the system. (C) The number of solid-like H2O molecules as determined by their mean-squared displacement with respect to averaged molecular positions. (D) The fraction of H2S molecules in the aqueous solution. The molecular configurations (I–IV) show the system evolution during the simulation, where the time index has been provided. H2O molecules are labeled as liquid-like (red spheres) and solid-like (green spheres), H2S molecules are represented as yellow spheres, and hydrogen bonds between solid-like H2O molecules are represented by white lines. We note that the spherical H2S nano-bubble cannot be clearly seen here since it is obscured by H2O molecules. | ||
Fig. 2 shows the average coordination numbers for first and second neighbors of H2S molecules for the same trajectory as in Fig. 1. Those H2S molecules within a distance of 5 Å of the central H2S molecule were defined as first (nearest) neighbors, while those within a distance of 5.5–7.5 Å were defined as second neighbors. We can see that as the simulation progresses, and particularly during nucleation, the average number of first neighbors decreases while the number of second neighbors increases (also see ESI†). The change of the coordination number, especially that of the second neighbors, as a function of time appears strongly correlated to that of the four order parameters presented in Fig. 1, indicating the important role of the second gas neighbors (i.e., solvent-separated gas molecules22,23,32) during the nucleation process of gas hydrates. Initially, small 512 cages are occasionally observed to form within the aqueous phase. These small cages tend to form around those H2S molecules with a high degree of second-neighbor H2S coordination. Fig. 2B shows an example of the formation of a small 512 cage around a H2S molecule with 12 second gas neighbors at about 18 ns. At this stage in the simulation the average number of second neighbors for H2S molecules in the solution is less than 4. Prior to nucleation such cages are transient and typically dissociate after a few hundred picoseconds, emphasizing the stochastic nature of the nucleation under the present simulation conditions. Those observations are consistent with previous observations for CH4 hydrate nucleation,18,20,21 and nucleation of coarse-grained model hydrates.22,23
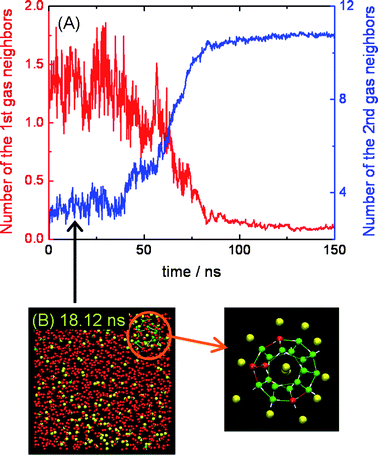 | ||
| Fig. 2 (A) shows the average number of first (red line) and second (blue line) neighbors (see text) for H2S molecules for the trajectory shown in Fig. 1. Molecular configuration (B) shows an example of formation of a small 512 cage (see expanded view) around a H2S molecule with 12 second gas neighbors at about 18 ns. The molecules are represented and colored as in Fig. 1. | ||
The hydrate nucleation event for this particular run of sys-A occurs at about 57 ns (see Fig. 1 and 2), where the conversion of the system from liquid- to solid-like behavior (i.e., crystal nucleation and growth) is completed within about 25 ns. The amorphous solid resulting at this stage exhibits short-ranged order but essentially no long-ranged structure. It includes components of structure I (sI), structure II (sII) and structure HS-I2,3hydrates; several such fragments are presented in Fig. 3. We also observe some unusual arrangements of regular sI, sII and HS-I cages, as well as other irregular cages (see ESI†). This amorphous structure is expected to evolve into a crystalline hydrate on a time scale beyond current MD simulations. However annealing of the amorphous solid at an elevated temperature (265 K) leads to a more recognizable crystalline structure within 40 ns (see ESI†). A recent study by Molinero and co-workers33 demonstrated that crystalline hydrates can grow out of an amorphous seed, which implies that macroscopic gas hydrates crystals may be proceeded by amorphous intermediate structures. This apparent two-step mechanism of the nucleation is consistent with observed behavior for CH4 hydrates,21 coarse-grained model hydrates with guests of different sizes and solubilities,22,23 as well as proteins,34–36 colloids,37 and CaCO3.38 We note that the formation of order through disordered intermediate solid-like structures has also been observed in our heterogeneous crystal growth simulations.30
 | ||
| Fig. 3 Images of several fragments of hydrate structure identified in the amorphous solid shown in Fig. 1(IV). (A), (B), and (C) show fragments of regular sI, sII, and HS-I structures (see text), respectively. (D) shows a transition structure from sI to sII, i.e., the connection of a 51262 cage and a 51264 cage by a 51263 cage. The H2O oxygens are represented by green spheres and white sticks are the hydrogens. The H2S molecules are represented by yellow spheres. | ||
One of the most interesting aspect(s) of the nucleation behavior observed here is the very short induction time for a two-phase H2S/H2O system at relatively low driving forces. Fig. 4 tracks F4 order parameter results for MD simulation runs for sys-A with temperatures ranging from ΔT = −40 to −60 K (i.e, roughly 14–20% undercoolings). To confirm the reproducibility of the nucleation events and to obtain estimates of average behavior, we have performed five independent simulations at each temperature. It is evident from Fig. 4 that the induction time decreases dramatically with decreasing temperature. We note that the runs at ΔT = −40 K were extended to 250 ns and a total of four trajectories were observed to nucleate (see ESI†). At an undercooling of −60 K, the system exhibits onset of nucleation behavior almost immediately (i.e., there is essentially no induction time). We conjecture that this rapid nucleation is related to the relatively high solubility of H2S in the aqueous phase, which for sys-A achieves about 45% of the fully occupied hydrate composition. We remark that this H2S concentration in solution is comparable to that necessary to achieve rapid nucleation of CH4 hydrates.21 Additionally, the observed nucleation behavior appeared unchanged by a pressure reduction to 0.5 MPa (see ESI†).
 | ||
| Fig. 4 Evolution of the F4 structural order parameters for sets of parallel nucleation simulations of sys-A at different undercoolings ranging from ΔT = −40 to −60 K (see legends). The inset in (E) shows the evolution of this order parameter during the first nanosecond of this trajectory, emphasizing the very rapid apparent onset of nucleation behavior at this temperature. | ||
With a lower H2S concentration in solution, about 30% of the fully occupied hydrate composition, sys-B (with a cylindrical H2S nano-bubble) tended to nucleate with longer induction times relative to sys-A at the same undercoolings. Fig. 5 compares typical nucleation behavior for sys-A and sys-B at ΔT = −50 K, where it is evident that sys-A nucleates within about 10 ns while sys-B requires about 100 ns. Molecular configurations taken from various stages of the simulation of sys-B are also presented in Fig. 5B, from which we can see that the nucleation behavior of sys-B is otherwise very similar to that of sys-A.
 | ||
| Fig. 5 (A) shows the evolution of the F4 structural order parameters for nucleation simulations of sys-A (with spherical H2S nano-bubble) and sys-B (with cylindrical H2S nano-bubble) at ΔT = −50 K. (B) shows several molecular configurations of sys-B during its simulation trajectory, where (I–IV) are labeled in (A). The molecules are represented and colored as in Fig. 1. The views in (B) are along the cylinder axis of the liquid H2S bubble. | ||
As shown in Fig. 6, similar (almost) instantaneous nucleation behavior was also observed for sys-B at low temperatures. At an undercooling of −60 K, sys-B appears to begin to nucleate almost immediately. In comparison with sys-A (Fig. 4), the overall nucleation behavior of sys-B appears shifted to lower temperatures by about 5−10 K. This is apparently related to the lower H2S solution concentration of sys-B (Table 1). A more quantitative study of the relationship between the induction time and gas concentration in aqueous solution is clearly warranted. Sys-C with a H2S liquid slab (and a solution composition only slightly lower than that of sys-B, see Table 1) was observed to nucleate at an undercooling of −60 K on a 100 ns timescale. We observe nucleation events very close to the H2S/H2O interfaces for sys-C, while for sys-A and sys-B we found no preference for nucleation at the interface. Molinero and co-workers22 performed 12 independent simulations of their coarse-grained models hydrate and found no marked preference for nucleation at the interface. While mass transport39 may be playing a role in the nucleation behavior of sys-C, further investigations are needed to clarify possible differences between the systems.
 | ||
| Fig. 6 Evolution of the F4 structural order parameters for sys-B (with a cylindrical H2S nano-bubble) at different undercoolings ranging from ΔT = −40 to −80 K (see legends). | ||
The above observations support the claim that gas composition is a critical order parameter in determining induction times for the nucleation of H2S hydrates. Additionally, the coordination number of gas molecules in solution appears to be an important factor. Fig. 7A shows the distribution of numbers of second neighbors (i.e., within a distance of 5.5–7.5 Å) of H2S molecules in aqueous solution recorded in the initial metastable solution systems at various undercoolings for sys-A. The distributions appear relatively insensitive to temperature, and H2S molecules with a high degree of second-neighbor coordination, which tend to favor formation of hydrate cages (as note above),18,22,23 are found at all the temperatures studied. The apparent temperature independence of the distribution curves shown in Fig. 7A and the observed reduced induction time at lower temperatures suggest that at a lower temperature (Fig. 4) cage formation can occur with a lower degree of second-neighbor coordination.
 | ||
| Fig. 7 The distribution of second-neighbor coordination numbers for gas molecules of (A) sys-A and (B) sys-A, sys-C, and a CH4/H2O system with a comparable gas concentration to that of sys-A. These distribution of coordination numbers are measured within the initial metastable systems, typically averaged over at least 10 ns. The simulation temperatures (undercoolings) are indicated in the legends. The melting temperatures for the present model H2S and CH4 hydrate systems are roughly 290 K (ref. 25) and 275 K (ref. 40), respectively. | ||
A comparison of distributions of numbers of second neighbors of H2S molecules within sys-A and sys-C is presented in Fig. 7B. We can see that with its lower gas concentration, the probability of H2S molecules with a high degree of second-neighbor coordination in sys-C is clearly lower than that in sys-A. The probability of H2S molecules with more than 7 second neighbors within sys-A is roughly one order of magnitude larger than that within sys-C. We note that the distribution of numbers of second neighbors of H2S molecules within sys-B is similar to that within sys-C. These observations suggest that increasing the gas concentration helps dramatically increase the probability of gas molecules exhibiting a high degree of second-neighbor coordination, thereby enhancing the tendency of cage formation and reducing the (average) induction time for nucleation. In previous work,21 it was demonstrated that an elevated local concentration of CH4 can dramatically reduce the induction time for nucleation of a CH4 hydrate. Fig. 7B also shows the distribution of second-neighbor coordination numbers for CH4 molecules in an aqueous solution with a gas concentration comparable to sys-A. At such elevated CH4 compositions, these solutions can nucleate rapidly (further information can be found in ref. 21). The similarity of the distribution numbers of second neighbors for the two systems suggests that the local concentration is critical in the early stage behavior of gas hydrate nucleation. We note that a common feature of CH4 and H2S is that they can fit into both small and large cages of a sI hydrate. A somewhat different (slower) nucleation behavior might be expected for other highly soluble molecules (such as tetrahydrofuran) which will fit only into large cages.23
From our sets of simulation results (Fig. 4), we can roughly estimate the temperature dependence of the average rate of nucleation (see ESI†) of sys-A. In Fig. 8, we see that the nucleation rate of these H2S/H2O systems increases very rapidly with decreasing the temperature from ΔT = −40 to −50 K. Upon a further decrease of the temperature, the nucleation rate appears to begin to decrease because the dynamics (of formation of order) becomes dominant and slow, although the induction time has become negligibly small. The nucleation rates observed here are nearly three orders of magnitude higher than that observed by Walsh et al.20 for CH4 hydrate under comparable undercooled conditions.40 While factors such as relative solubilities and mobilities appear to contribute to this difference, further careful studies are needed to elucidate the various contributions to the overall rate. We remark that this very rapid nucleation for the H2S/H2O system is observed with undercoolings of only 15–20%. Even for simple Lennard-Jones systems, an undercooling of more than 25% is needed to observe nucleation on a typical MD timescale.41,42 For a simple molecular system such as CO2, an undercooling of 30% or more is needed to observe nucleation within current computational capabilities.43 In this context the present results represent the direct observation of nucleation under rather modest conditions. Hence H2S/H2O systems are computationally advantageous in exploring the nucleation behavior of a gas hydrate in comparison to CH4/H2O or other common gas hydrates. Moreover, the apparent high nucleation rate of H2S/H2O systems suggests that the H2S species may, at least under some conditions, play a key role in hydrate formation. For example, it may help initiate the formation of a hydrate crystal in a pipeline, and once formed, a CH4 hydrate can then easily continue to grow (perhaps causing a blockage in a pipeline). We remark that recent infrared spectroscopy experiments reported by Devlin and co-workers14,44 have suggested that the inclusion of H2S in aqueous solution can dramatically enhance the rate of a gas hydrate formation.
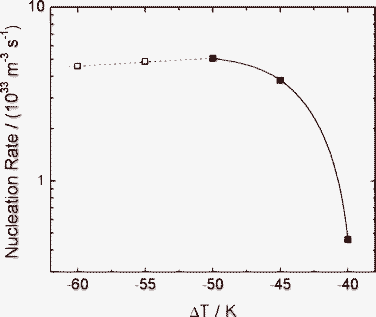 | ||
| Fig. 8 Nucleation rates of a two-phase H2S/H2O system with a spherical H2S nano-bubble (sys-A) as a function of undercooling. The solid squares and the red solid line represent the behavior prior to the maximum rate. The hollow squares and the red dashed line represent estimates at lower temperatures. The ice point of the TIP4P/2005 model is estimated to be 248 K (ΔT = −42 K, see ref. 26) at 50 MPa. | ||
4. Conclusions
Through MD simulations, this report demonstrates that with appropriately high local gas concentration, provided here by two-phase H2S/H2O systems, the nucleation of a gas hydrate can be extremely rapid (in a molecular simulation context) even at relatively modest undercoolings. We observe almost instantaneous and highly reproducible nucleation at only 15–20% undercoolings (−60 to −40 K). The nucleation process is characterized as two-step, with initial formation of an amorphous-like solid, consistent with observations for CH4 hydrates21 and coarse-grained model hydrates with guests of different sizes and solubilities,22,23 as well as for proteins,34–36 colloids,37 and CaCO3.38The present results illustrate that increasing the gas concentration helps increase the probability of gas molecules with a high degree of second-neighbor coordination, thereby reducing the (average) induction time for nucleation of a gas hydrate. CH4/H2O and H2S/H2O systems with a comparable gas concentrations show very similar distributions of numbers of second neighbors. The (absolute) local concentration can be identified as a critical factor in the early stage behavior of gas hydrate nucleation. The relatively high solubility of H2S molecules in aqueous solution appears to help promote nucleation and provides a computationally advantageous model system to investigate nucleation behavior of a gas hydrate. A more quantitative study of the relationship between the average induction time and gas concentration in aqueous solution clearly warrants further detailed study.
Acknowledgements
We are grateful for the financial support of the Natural Sciences and Engineering Research Council of Canada. We also acknowledge computational resources made available via WestGrid (http://www.westgrid.ca) and the University of Calgary.References
- E. D. Sloan and C. A. Koh, in Clathrate Hydrates of Natural Gases, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- J. Vatamanu and P. G. Kusalik, J. Am. Chem. Soc., 2006, 128, 15588 CrossRef CAS.
- L. Yang, C. A. Tulk, D. D. Klug, I. L. Moudrakovski, C. I. Ratcliffe, J. A. Ripmeester, B. C. Chakoumakos, L. Ehm, C. D. Martin and J. B. Parise, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6060 CrossRef CAS.
- G. A. Jeffrey, in Comprehensive Supramol Chem, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vogtle, and J. M. Lehn, Pergamon, Oxford, New York, 1996 Search PubMed.
- E. D. Sloan, Nature, 2003, 426, 353 CrossRef.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743 CrossRef CAS.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Science, 2004, 306, 469 CrossRef CAS.
- Y. Park, D. Y. Kim, J. W. Lee, D. G. Huh, K. P. Park, J. Lee and H. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12690 CrossRef CAS.
- J. Bohannon, Science, 2008, 319, 1753 CrossRef CAS.
- J. C. McElwain, J. Wade-Murphy and S. P. Hesselbo, Nature, 2005, 435, 479 CrossRef CAS.
- K. A. Kvenvolden, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3420 CrossRef.
- C. A. Koh, R. P. Wisbey, X. P. Wu, R. E. Westacott and A. K. Soper, J. Chem. Phys., 2000, 113, 6390 CrossRef CAS.
- Y. R. Shen and V. Ostroverkhov, Chem. Rev., 2006, 106, 1140 CrossRef CAS.
- J. P. Devlin and I. A. Monreal, Chem. Phys. Lett., 2010, 492, 1 CrossRef CAS.
- F. Lehmkuhler, M. Paulus, C. Sternemann, D. Lietz, F. Venturini, C. Gutt and M. Tolan, J. Am. Chem. Soc., 2009, 131, 585 CrossRef.
- H. Conrad, F. Lehmkuhler, C. Sternemann, A. Sakko, D. Paschek, L. Simonelli, S. Huotari, O. Feroughi, M. Tolan and K. Hamalainen, Phys. Rev. Lett., 2009, 103, 218301 CrossRef.
- C. Moon, P. C. Taylor and P. M. Rodger, J. Am. Chem. Soc., 2003, 125, 4706 CrossRef CAS.
- R. W. Hawtin, D. Quigley and P. M. Rodger, Phys. Chem. Chem. Phys., 2008, 10, 4853 RSC.
- J. F. Zhang, R. W. Hawtin, Y. Yang, E. Nakagava, M. Rivero, S. K. Choi and P. M. Rodger, J. Phys. Chem. B, 2008, 112, 10608 CrossRef CAS.
- M. R. Walsh, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Science, 2009, 326, 1095 CrossRef CAS.
- J. Vatamanu and P. G. Kusalik, Phys. Chem. Phys. Chem., 2010, 12, 15065 Search PubMed.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Am. Chem. Soc., 2010, 132, 11806 CrossRef CAS.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Phys. Chem. B, 2010, 114, 13796 CrossRef CAS.
- J. J. Carroll and Knovel, in Natural Gas Hydrates: A Guide for Engineers, Gulf Professional Publishing, Houston, TX, 2003 Search PubMed.
- S. Liang and P. G. Kusalik, J. Phys. Chem. B, 2010, 114, 9563 CrossRef CAS.
- T. R. Forester, I. R. McDonald and M. L. Klein, Chem. Phys., 1989, 129, 225 CrossRef CAS.
- K. Gillis, J. Vatamanu, M. S. G. Razul, and P. G. Kusalik, in Applied Parallel Computing – State of the Art in Scientific Computing, ed. B. Kagstrom, E. Elmroth, J. Dongarra and J. Wasniewski, Springer-Verlag, Berlin, 2007 Search PubMed.
- M. S. G. Razul, J. G. Hendry and P. G. Kusalik, J. Chem. Phys., 2005, 123, 204722 CrossRef.
- J. Vatamanu and P. G. Kusalik, J. Chem. Phys., 2007, 126, 124703 CrossRef CAS.
- S. Liang and P. G. Kusalik, Chem. Phys. Lett., 2010, 494, 123 CrossRef CAS.
- P. M. Rodger, T. R. Forester and W. Smith, Fluid Phase Equilib., 1996, 116, 326 CrossRef CAS.
- M. Matsumoto, J. Phys. Chem. Lett., 2010, 1, 1552 Search PubMed.
- L. C. Jacobson and V. Molinero, J. Am. Chem. Soc., 2011 Search PubMed , to be published.
- P. R. tenWolde and D. Frenkel, Science, 1997, 277, 1975 CrossRef.
- H. J. Liu, S. K. Kumar and J. F. Douglas, Phys. Rev. Lett., 2009, 103, 018101 CrossRef.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621 CrossRef CAS.
- J. R. Savage and A. D. Dinsmore, Phys. Rev. Lett., 2009, 102, 198302 CrossRef CAS.
- E. M. Pouget, P. H. H. Bomans, J. Goos, P. M. Frederik, G. de With and N. Sommerdijk, Science, 2009, 323, 1455 CrossRef CAS.
- S. Liang and P. G. Kusalik, J. Am. Chem. Soc., 2011, 133, 1870 CrossRef CAS.
- M. M. Conde and C. Vega, J. Chem. Phys., 2010, 133, 064507 CrossRef CAS.
- C. Desgranges and J. Delhommelle, Phys. Rev. Lett., 2007, 98, 235502 CrossRef.
- C. Desgranges and J. Delhommelle, J. Am. Chem. Soc., 2006, 128, 10368 CrossRef CAS.
- J. M. Leyssale, J. Delhommelle and C. Millot, J. Chem. Phys., 2005, 122, 184518 CrossRef.
- V. Buch, J. P. Devlin, I. A. Monreal, B. Jagoda-Cwiklik, N. Uras-Aytemiz and L. Cwiklik, Phys. Chem. Chem. Phys., 2009, 11, 10245 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00021g |
| This journal is © The Royal Society of Chemistry 2011 |