DOI:
10.1039/C1SC00122A
(Edge Article)
Chem. Sci., 2011,
2, 1273-1277
Asymmetric organocatalytic [3 + 2]-annulation strategy for the synthesis of N-fused heteroaromatic compounds†
Received
2nd March 2011
, Accepted 31st March 2011
First published on 19th April 2011
Abstract
Hydroxyalkyl- or aminoalkyl-substituted N-fused heteroaromatic compounds can be efficiently accessed via an organocatalytic [3 + 2]-annulation strategy. The developed cascades proceed in a highly enantioselective manner and benefit from broad substrate scope, operational simplicity, easily available starting materials as well as low catalyst loadings.
Introduction
The chemistry and biological properties of nitrogen-containing heteroaromatic molecules constitute a very rapidly developing field of research due to their presence in numerous naturally occurring compounds, widespread applications in organic synthesis and diverse biological activity.1
Among the multitude of N-heteroaromatic compounds N-fused derivatives such as indolizines and azaindolizines occupy a prominent position (Fig. 1).2 In particular, the biological activity of imidazo[1,2-a]pyridines is well recognized.3–5 For instance, they encompass a broad spectrum of antiviral, cytotoxic, antibacterial, fungicidal, anti-inflammatory properties3 and are bradykinin B2-receptor as well as CXC-chemokine receptor 4 (CXCR4) antagonists.4 Moreover, these compounds exhibit inhibitory activity against cyclin dependant kinase 2 (CDK2) and posses high affinity for GABA, and type I benzodiazepine receptors.5 For these reasons these compounds are considered privileged scaffolds for drug discovery. This is supported by imidazo[1,2-a]pyridine derivatives such as Zolpidem (hypnotic properties) and Alpidem (anxiolytic properties) which are commercially available drugs.6 Furthermore, the optically active GSK812397, with a hydroxymethyl substituent in the 3-position, is a candidate for application in HIV infection treatment.4a,b As a consequence, the development of methods allowing for efficient and facile construction of imidazo[1,2-a]pyridine and indolizine rings is an important goal in modern organic chemistry that has received considerable attention over the years.7,8 Noteworthily, to the best of our knowledge, no enantioselective methods leading to the formation of optically active hydroxyalkyl- or aminoalkyl-substituted imidazo[1,2-a]pyridines and indolizines have been disclosed in the literature so far. Given the importance of these N-fused heteroaromatic derivatives and their potential biological properties, the development of new methodologies for their preparation in an enantioselective fashion is of great importance for organic and medicinal chemistry. In the following, we will provide efficient asymmetric access to these valuable heteroaromatic scaffolds applying an organocatalytic9 [3 + 2]-annulation strategy as presented in Scheme 1. We will report that readily available α,β-unsaturated aldehydes, 2-aminopyridines and ethyl 2-pyridylacetate can be utilized in highly enantioselective one-pot reaction cascades10 offering a facile and efficient entry to optically active hydroxyalkyl- and aminoalkyl-substituted imidazo[1,2-a]pyridines and indolizines.
![Asymmetric organocatalytic [3 + 2]-annulation strategy for the synthesis of optically active N-fused heteroaromatic compounds.](/image/article/2011/SC/c1sc00122a/c1sc00122a-s1.gif) |
| | Scheme 1 Asymmetric organocatalytic [3 + 2]-annulation strategy for the synthesis of optically active N-fused heteroaromatic compounds. | |
Results and discussion
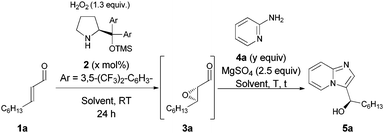
The optimization studies were initiated using trans-2-nonenal 1a and 2-aminopyridine 4a as model substrates (Table 1). Organocatalytic epoxidation of 1a was terminated within 24 h and to our delight, the applied protocol11 was fully compatible with the annulation step yielding the desired 3-hydroxyalkylimidazo[1,2-a]pyridine 5a. However, a large amount of unreacted 4a was present in the crude reaction mixture implying that decomposition of the intermediating 2,3-epoxy aldehyde 3a occurred under the reaction conditions. The result was improved by rising the reaction temperature to 40 °C. On the contrary, increase of the amount of 4a led to even faster decomposition of 3a resulting in lower overall conversion. Disappointingly, all attempts to isolate pure product 5a and separate it from unreacted 4a proved unsuccessful (Table 1, entries 1–3). Further studies revealed that 5a can be obtained in pure form by decreasing the amount of 4a used in the annulation step (Table 1, entries 4, 5). Moreover, the use of toluene as the solvent had a beneficial influence on the cascade outcome, albeit higher catalyst loading (5 mol%) was required in order to accomplish the epoxidation of 1a within 24 h. Performing the annulation step at 60 °C for 1 h using 0.8 equiv. of 4a resulted in the formation of imidazo[1,2-a]pyridine 5a in 58% yield and 94% ee (Table 1, entry 8). Noteworthily, the overall yields of the one-pot reaction cascade are given, which are not based on the limiting factor4a.
Table 1 Enantioselective synthesis of 3-hydroxyalkyl imidazo[1,2-a]pyridines—optimization studiesa
| |
Solvent (cat. loading) |
4a (equiv) |
T (°C)b |
t (h)c |
Conv. (%)d |
ee (%)e |
|
Reactions performed on 0.2 mmol scale in 0.4 mL of the solvent (for details see Supporting Information†).
Reaction temperature for the 2nd step.
Reaction time for the 2nd step.
Conversion of 4a in the 2nd step as determined by 1H NMR spectroscopy. In parentheses the yield of isolated product 5a is given.
Determined by chiral stationary phase HPLC.
Reaction performed in absence of MgSO4 in the 2nd step.
|
| 1 |
DCM (2.5) |
1 |
RT |
20 |
55 (nd) |
nd |
| 2 |
DCM (2.5) |
1 |
40 |
20 |
66 (nd) |
nd |
| 3 |
DCM (2.5) |
1.2 |
40 |
20 |
44 (nd) |
nd |
| 4 |
DCM (2.5) |
0.8 |
40 |
20 |
90 (54) |
92 |
| 5 |
DCM (2.5) |
0.7 |
40 |
20 |
>95 (52) |
92 |
| 6 |
Toluene (5) |
0.9 |
60 |
1 |
87 (nd) |
nd |
| 7 |
Toluene (5) |
0.8 |
60 |
1 |
>95 (57) |
94 |
| 8f |
Toluene (5) |
0.8 |
60 |
1 |
>95 (58) |
94 |
| 9f |
Toluene (5) |
0.7 |
60 |
1 |
>95 (55) |
94 |
With the optimized reaction conditions in hand, we turned our attention to the substrate scope (Table 2). To our delight, various α,β-unsaturated aldehydes could participate in the developed one-pot reaction sequence. However, at this stage it was found that in most cases, the amount of 4a had to be further reduced to 0.7 equiv. in order to obtain pure 5. Importantly, the scope of the reaction was very broad. Various linear and γ-branched aliphatic aldehydes could be utilized in the developed one-pot reaction cascade (Table 2, entries 1–5). Furthermore, different functional groups in the side-chain of 1 were also well tolerated (Table 2, entries 6–9). However, cinnamaldehyde and its derivatives did not give satisfactory results under these reaction conditions.
Table 2 Enantioselective synthesis of 3-hydroxyalkyl imidazo[1,2-a]pyridines—aldehyde scopea
| Entry |
1 (R) |
Yield (%)b |
ee (%)c |
|
Reactions performed on 0.2 mmol scale in 0.4 mL toluene (see Supporting Information for details†).
Overall yield given.
Determined by chiral stationary phase HPLC.
Performed using 0.8 equiv. of 4a in the annulation step.
Performed using 10 mol% of the catalyst 2.
Performed using 0.6 equiv. of 4a in the annulation step.
|
| 1d |
1a (Hex) |
5a—58 |
94 |
| 2 |
1b (Pen) |
5b—52 |
95 |
| 3 |
1c (Pr) |
5c—54 |
92 |
| 4 |
1d (iPr) |
5d—54 |
98 |
| 5e |
1e (Me) |
5e—31 |
91 |
| 6e |
1f (E-Hex-3-enyl) |
5f—52 |
90 |
| 7f |
1g (Z-Hex-3-enyl) |
5g—37 |
94 |
| 8 |
1h (CH2CH2Ph) |
5h—52 |
94 |
| 9 |
1i (CH2OBn) |
5i—41 |
92 |
With the synthesis of 3-hydroxyalkyl imidazo[1,2-a]pyridines 5 being accomplished, we undertook studies on the development of a one-pot reaction cascade leading to the formation of 3-aminoalkyl derivatives using 2,3-aziridine aldehydes as key intermediates.11 The optimization studies revealed (see Supporting Information for details†) that the use of toluene as a solvent and 0.9 equiv. of 4a at 60 °C ensures the best efficiency of the one-pot reaction cascade. Under the optimal reaction conditions a variety of α,β-unsaturated aldehydes 1a–i reacted smoothly (Table 3) to give the target products 6a–i in good yields (53–68% yield) and with excellent enantiomeric excesses (92–97% ee).
Table 3 Enantioselective synthesis of 3-aminoalkyl imidazo[1,2-a]pyridines—aldehyde scopea
| Entry |
1 (R) |
Yield (%)b |
ee (%)c |
|
Reactions performed on 0.1 mmol scale in 0.5 mL toluene (see Supporting Information for details†).
Overall yield given.
Determined by chiral stationary phase HPLC.
|
| 1 |
1a (Hex) |
6a—68 |
97 |
| 2 |
1b (Pen) |
6b—58 |
92 |
| 3 |
1c (Pr) |
6c—64 |
96 |
| 4 |
1d (iPr) |
6d—62 |
96 |
| 5 |
1e (Me) |
6e—53 |
96 |
| 6 |
1f (E-Hex-3-enyl) |
6f—60 |
94 |
| 7 |
1g (Z-Hex-3-enyl) |
6g—60 |
95 |
| 8 |
1h (CH2CH2Ph) |
6h—60 |
96 |
| 9 |
1i (CH2OBn) |
6i—61 |
95 |
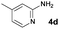
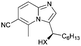
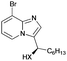
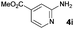
In the course of the further studies, the possibility to employ diversely substituted 2-aminopyridines 4b–i in the developed reaction sequence was evaluated (Table 4). To our delight, both developed one-pot reaction cascades proved to be general, since 2-aminopyridines bearing either electron-donating (Table 4, entries 1–6) or electron-withdrawing substituents (Table 4, entries 9–17) reacted effectively to afford the target products in moderate to good overall yields (40–83%) and excellent enantioselectivities (90–98% ee). However, in some of the cases the amount of 2-aminopyridine 4 used had to be re-optimized, in order to avoid unreacted 4 in the crude reaction mixtures or to increase the conversion of 3. It is also worth noting that the position of the substituent on the pyridine ring had no significant influence on the cascade outcome. Furthermore, disubstituted 2-aminopyridine 4e could also be successfully applied in both reaction sequences, although with slightly lower yields (Table 4, entries 7, 8). The usefulness and practicality of the developed approach was further demonstrated in a gram-scale synthesis of 5o (Table 4, entry 12). The reaction cascade was performed on 10 mmol scale and proceeded efficiently yielding 5o in good yield and in a highly enantioselective manner.
Table 4 Enantioselective synthesis of 3-hydroxyalkyl and 3-aminoalkyl imidazo[1,2-a]pyridines—2-aminopyridine scope
| |
4
|
5 or 6 |
X |
Yield (%)c |
ee (%)d |
|
Reaction performed on 0.2 mmol scale in 0.4 mL toluene using 0.7 equiv. of 4 in the annulation step (see Supporting Information for details†).
Reaction performed on 0.1 mmol scale in 0.5 mL toluene using 0.9 equiv. of 4 in the annulation step (see Supporting Information for details†).
Overall yield given.
Determined by chiral stationary phase HPLC.
Performed using 0.6 equiv. of 4 in the annulation step.
Performed using 0.9 equiv. of 4 in the annulation step.
Performed on 10 mmole scale using 0.9 equiv. of 4 in the annulation step.
Annulation step performed with 0.8 equiv. of 4 for 90 min.
Performed using 0.8 equiv. of 4 in the annulation step.
|
| 1a |

|

|
O |
5j—40 |
90 |
| 2b |
NTs |
6j—68 |
91 |
| 3a |

|

|
O |
5k—60 |
90 |
| 4b |
NTs |
6k—83 |
97 |
| 5a,e |
|

|
O |
5l—45 |
94 |
| 6b |
NTs |
6l—63 |
96 |
| 7a,e |

|
|
O |
5m—36 |
91 |
| 8b |
NTs |
6m—51 |
98 |
| 9a,f |

|

|
O |
5n—47 |
91 |
| 10b |
NTs |
6n—64 |
94 |
| 11a,f |

|

|
O |
5o—44 |
96 |
| 12g |
O |
5o—48 |
98 |
| 13b |
NTs |
6o—50 |
92 |
| 14a |

|
|
O |
5p—44 |
92 |
| 15b,e |
NTs |
6p—56 |
97 |
| 16a,h |
|

|
O |
5q—52 |
94 |
| 17b,i |
NTs |
6q—63 |
95 |
In order to annulate an indolizine ring, ethyl 2-pyridylacetate 7 was utilized in the annulation step, which further expanded the scope of the methodology (Scheme 2). Both 2,3-epoxy and 2,3-aziridine aldehydes derived from trans-2-nonenal 1a reacted smoothly with 7 under the previously optimized one-pot conditions. Disappointingly, the hydroxyalkyl derivative 8a turned out to be unstable and decomposed during isolation. On the contrary, indolizine derivative 8b was perfectly stable and could be isolated by FC in 64% yield and 97% ee.
![Asymmetric organocatalytic [3 + 2]-annulation strategy for the synthesis of indolizine derivatives.](/image/article/2011/SC/c1sc00122a/c1sc00122a-s2.gif) |
| | Scheme 2 Asymmetric organocatalytic [3 + 2]-annulation strategy for the synthesis of indolizine derivatives. | |
The bromo-substituted imidazo[1,2-a]pyridines can serve as versatile synthons enabling introduction of various functional groups onto the imidazo[1,2-a]pyridine ring and expanding the availability of the target optically active products as demonstrated in the synthesis of 5r and 5s (Scheme 3). It was shown that the bromine atom in 5o could easily be replaced by applying 5o in standard palladium-catalyzed cross-coupling reactions. In the first attempt, a Sonogashira coupling was performed allowing for introduction of an alkynyl substituent into the target molecule 5r. Likewise, a Suzuki coupling with phenylboronic acid afforded 5s in high yield. Importantly, despite the basic reaction conditions and prolonged heating at elevated temperature, no significant racemization took place and the final products 5r,s were obtained in high enantiomeric excesses. These results clearly indicate high configurational stability of the imidazo[1,2-a]pyridines synthesized.
 |
| | Scheme 3 Applications of 5o in Pd-catalyzed cross-coupling reactions. Reagents and conditions: a) ethynyltrimethylsilane (3 equiv.), PdCl2(PPh3)2 (5 mol%), CuI (5 mol%), Et3N, 60 °C, overnight. b) PhB(OH)2 (3 equiv.), PdCl2(PPh3)2 (2.5 mol%), 2M aq. K2CO3, toluene, 60 °C, overnight. | |
The proposed mechanism for the developed one-pot reaction cascades is depicted in Scheme 4. The key intermediates 2,3-epoxy- or 2,3-aziridine aldehydes 3 or 14 are generated in a highly enantioselective fashion via a catalytic cycle proceeding by the formation of iminium ion 9 and enamines 10 or 11 as reactive intermediates responsible for the enantio- and diastereoselectivity of the process. Intermediates 3 or 14 are in turn reacted with 2-aminopyridines 4 or ethyl 2-pyridylacetate 7. The reaction presumably proceeds via the formation of hemiaminals 15 or 16 followed by intramolecular epoxide or aziridine ring-opening via the pyridine nitrogen atom. Subsequent dehydration of 17 or 18 to give 19 or 20 correspondingly and aromatization thereof yields target products 5, 6 or 8.
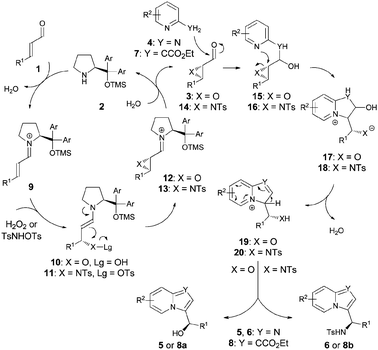 |
| | Scheme 4 Mechanistic proposal for the one-pot reaction cascades. | |
The absolute configuration of the products was assigned to be R for the product 5a–h,j–s, 6a–h,j–s and 8b and S for the products 5i and 6i by analogy to previous results on epoxidations of α,β-unsaturated aldehydes 1 catalyzed by 2 and their applications in target-oriented syntheses.11a–d The correctness of these configurational assignments was unambiguously confirmed by single crystal X-ray analysis of 6c (Fig. 2).12
Conclusion
In conclusion, an efficient enantioselective organocatalytic [3 + 2]-annulation strategy for the preparation of imidazo[1,2-a]pyridine and indolizine derivatives has been established. The presented methodology benefits from the high enantioselectivities obtained, broad substrate scope, operational simplicity, easily available starting materials as well as low catalyst loadings. The possibility to perform the developed cascade on gram-scale and to apply the products in palladium-catalyzed cross-coupling reactions was also demonstrated. We believe that the presented methodology opens access to these interesting compounds which are now available for biological evaluation.
Acknowledgements
This work was made possible by grants from OChemSchool, Carlsberg Foundation, FNU and scholarship from Foundation for Polish Science (Kolumb Programme - ŁA). We thank Dr. Jacob Overgaard for performing X-ray analysis.
Notes and references
-
(a)
J. A. Joule and K. Mills, Heterocyclic Chemistry, 4th ed.; Blackwell: Oxford, U.K., 2000 Search PubMed;
(b)
Comprehensive Heterocyclic Chemistry: The Structure, Reactions, Synthesis, and Uses of Heterocyclic Compounds, Vols. 1–8, (ed.: A. R. Katrizky and C. W. Rees), ed.; Pergamon Press: Oxford, U.K., 1984 Search PubMed;
(c) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS.
-
(a)
A. S. Howard, in Comprehensive Heterocyclic Chemistry II, Vol. 8, (ed.: A. R. Katritzky, C. W. Rees and E. F. V. Scriven), Pergamon Press: Oxford, U.K., 1996, pp. 249–286 Search PubMed;
(b)
G. Jones, in Comprehensive Heterocyclic Chemistry II, Vol. 8, (ed.: A. R. Katritzky, C. W. Rees and E. F. V. Scriven), Pergamon Press: Oxford, U.K., 1996, pp. 287–343 Search PubMed;
(c)
F. Couty and G. Evano, in Comprehensive Heterocyclic Chemistry III, Vol. 11, (ed.: A. R. Katritzky, A. R. Ramsden, E. F. V. Scriven and R. J. K. Taylor), Elsevier: Oxford, 2008, pp. 409–499 Search PubMed.
-
(a) M. Lhassani, O. Chavignon, J.-M. Chezal, J.-C. Teulade, J.-P. Chapat, R. Snoeck, G. Andrei, J. Balzarini, E. De Clercq and A. Gueiffier, Eur. J. Med. Chem., 1999, 34, 271 CrossRef CAS;
(b) J. B. Veron, C. Enguehard-Gueiffier, R. Snoeck, G. Andrei, E. DeClercq and A. Gueiffier, Bioorg. Med. Chem., 2008, 16, 9536 CrossRef CAS;
(c) K. S. Gudmundsson, J. D. Williams, J. C. Drachs and L. B. Townsend, J. Med. Chem., 2003, 46, 1449 CrossRef CAS;
(d) M. A. Vilchis-Reyes, A. Zentella, M. A. Martínez-Urbina, Á. Guzmán, O. Vargas, M. T. Ramírez Apan, J. L. Ventura Gallegos and E. Díaz, Eur. J. Med. Chem., 2010, 45, 379 CrossRef CAS;
(e) Y. Rival, G. Grassy and G. Michel, Chem. Pharm. Bull., 1992, 40, 1170 CAS;
(f) M. H. Fisher and A. Lusi, J. Med. Chem., 1972, 15, 982 CrossRef CAS;
(g) Y. Rival, G. Grassy, A. Taudou and R. Ecalle, Eur. J. Med. Chem., 1991, 26, 13 CrossRef CAS;
(h) K. C. Rupert, J. R. Henry, J. H. Dodd, S. A. Wadsworth, D. E. Cavender, G. C. Olini, B. Fahmy and J. J. Siekierka, Bioorg. Med. Chem. Lett., 2003, 13, 347 CrossRef CAS.
-
(a)
K. Gudmundsson and S. D. Boggs, PCT Int. Appl. WO 2006026703, 2006; CAN 2006, 144, p. 274;
(b) S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781 Search PubMed;
(c) Y. Abe, H. Kayakiri, S. Satoh, T. Inoue, Y. Sawada, K. Imai, N. Inamura, M. Asano, C. Hatori, A. Katayama, T. Oku and H. Tanaka, J. Med. Chem., 1998, 41, 564 CrossRef CAS.
-
(a) M. Anderson, J. F. Beattie, G. A. Breault, J. Breed, K. F. Byth, J. D. Culshaw, R. P. Ellston, S. Green, C. A. Minshull, R. A. Norman, R. A. Pauptit, J. Stanway, A. P. Thomas and P. J. Jewsbury, Bioorg. Med. Chem. Lett., 2003, 13, 3021 CrossRef CAS;
(b) K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes and A. P. Thomas, Bioorg. Med. Chem. Lett., 2004, 14, 2245 CrossRef CAS;
(c) C. Hamdouchi, B. Zhong, J. Mendoza, E. Collins, C. Jaramillo, J. E. De Diego, D. Robertson, C. D. Spencer, B. D. Anderson, S. A. Watkins, F. Zhanga and H. B. Brooks, Bioorg. Med. Chem. Lett., 2005, 15, 1943 CrossRef CAS;
(d) S. C. Goodacre, L. J. Street, D. J. Hallett, J. M. Crawforth, S. Kelly, A. P. Owens, W. P. Blackaby, R. T. Lewis, J. Stanley, A. J. Smith, P. Ferris, B. Sohal, S. M. Cook, A. Pike, N. Brown, K. A. Wafford, G. Marshall, J. L. Castro and J. R. Atack, J. Med. Chem., 2006, 49, 35 CrossRef CAS;
(e) G. Trapani, M. Franco, L. Ricciardi, A. Latrofa, G. Genchi, E. Sanna, F. Tuveri, E. Cagetti, G. Biggio and G. Liso, J. Med. Chem., 1997, 40, 3109 CrossRef CAS;
(f) G. Trapani, M. Franco, A. Latrofa, L. Ricciardi, A. Carotti, M. Serra, E. Sanna, G. Biggio and G. Liso, J. Med. Chem., 1999, 42, 3934 CrossRef CAS.
-
(a) T. S. Harrison and G. M. Keating, CNS Drugs, 2005, 19, 65 CrossRef CAS;
(b) G. Mereu, G. Carcangiu, A. Concas, N. Passino and G. Biggio, Eur. J. Pharmacol., 1990, 179, 339 CrossRef CAS;
(c) M. Anzini, A. Cappelli, S. Vomero, G. Giorgi, T. Langer, G. Bruni, R. Romeo and A. S. Basile, J. Med. Chem., 1996, 39, 4275 CrossRef CAS.
- For reviews, see:
(a) C. Hulme and Y.-S. Lee, Mol. Diversity, 2008, 12, 1 CrossRef CAS;
(b) M. Shipman, Sci. Synth., 2001, 10, 745 Search PubMed; for selected examples, see:
(c) Z. Bahlaouan, M. Abarbri, A. Duchêne, J. Thibonnet, N. Henry, C. Enguehard-Gueiffier and A. Gueiffier, Org. Biomol. Chem., 2011, 9, 1212 RSC;
(d) K. C. Chunavala, G. Joshi, E. Suresh and S. Adimurthy, Synthesis, 2011, 635 CAS;
(e) Q. Liao, L. Zhang, S. Li and C. Xi, Org. Lett., 2011, 13, 228 CrossRef CAS;
(f) J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200 CrossRef CAS;
(g) A. L. Rousseau, P. Matlaba and C. J. Parkinson, Tetrahedron Lett., 2007, 48, 4079 CrossRef CAS.
- For selected examples of synthesis of racemic hydroxyalkyl- or aminoalkyl-substituted imidazo[1,2-a]pyridines or indolizines, see:
(a) I. A. Zamkova, O. O. Chekotylo, O. V. Geraschenko, O. O. Grygorenko, P. K. Mykhailiuk and A. A. Tolmachev, Synthesis, 2010, 1692 CAS;
(b) P. C. Lima, M. A. Avery, B. L. Tekwani, H.de M. Alves, E. J. Barreiro and C. A. M. Fraga, Farmaco, 2002, 57, 825 CrossRef CAS;
(c) C. Burkholder, W. R. Dolbier Jr., M. Médebielle and S. Ait-Mohand, Tetrahedron Lett., 2001, 42, 3459 CrossRef CAS;
(d) Y. Miki, Y. Hiroishi, H. Hachiken and S. Takemura, J. Heterocycl. Chem., 1991, 28, 45 CrossRef CAS;
(e) E. Öhler, M. El-Badawi and E. Zbiral, Chem. Ber., 1985, 118, 4099 CrossRef;
(f) E. Öhler, E. Zbiral and M. El-Badawi, Tetrahedron Lett., 1983, 24, 5599 CrossRef;
(g) W. C. Lumma Jr. and J. P. Springer, J. Org. Chem., 1981, 46, 3735 CrossRef.
- For recent reviews on organocatalysis, see:
(a) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS;
(b) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Chem. Rev., 2007, 107, 5413 CrossRef CAS (special issue on organocatalysis);
(c) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS;
(d) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef;
(e) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS;
(f) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC;
(g) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jørgensen, Chem. Commun., 2011, 47, 632 RSC;
(h)
Enantioselective Organocatalysis, (Ed.: P. I. Dalko), Wiley-VCH, Weinheim, 2007 Search PubMed.
- For selected examples of organocatalytic one-pot reaction cascades, see:
(a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS;
(b) B. Westermann, M. Ayaz and S. S. van Berkel, Angew. Chem., Int. Ed., 2010, 49, 846 CAS;
(c) H. Ishikawa, M. Honma and Y. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 2824 CrossRef CAS;
(d) G. Dickmeiss, K. L. Jensen, D. Worgull, P. T. Franke and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 1580 CrossRef CAS;
(e) N. T. Jui, E. C. Y. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10015 CrossRef CAS;
(f) S. Belot, K. A. Vogt, C. Besnard, N. Krause and A. Alexakis, Angew. Chem., Int. Ed., 2009, 48, 8923 CrossRef CAS;
(g) H. Jiang, N. Holub, M. W. Paixão, C. Tiberi, A. Falcicchio and K. A. Jørgensen, Chem.–Eur. J., 2009, 15, 9638 CrossRef CAS;
(h) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS;
(i) D. Enders, M. R. M. Hüttl, J. Runsink, G. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 46, 467 CrossRef CAS;
(j) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS.
-
(a) Ł. Albrecht, H. Jiang, G. Dickmeiss, B. Gschwend, S. G. Hansen and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 9188 CrossRef CAS;
(b) H. Jiang, B. Gschwend, Ł. Albrecht and K. A. Jørgensen, Org. Lett., 2010, 12, 5052 CrossRef CAS;
(c) Ł. Albrecht, L. K. Ransborg, B. Gschwend and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 17886 CrossRef CAS; for the first contribution, see:
(d) M. Marigo, J. Franzén, T. B. Poulsen, Z. Wei and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 6964 CrossRef CASfor other examples, see:
(e) H. Sundén, I. Ibrahem and A. Córdova, Tetrahedron Lett., 2006, 47, 99 CrossRef CAS;
(f) S. Lee and D. W. C. MacMillan, Tetrahedron, 2006, 62, 11413 CrossRef CAS;
(g) J. Vesely, I. Ibrahem, G.-L. Zhao, R. Rios and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 778 CrossRef CAS;
(h) H. Arai, N. Sugaya, N. Sasaki, K. Makino, S. Lectard and Y. Hamada, Tetrahedron Lett., 2009, 50, 3329 CrossRef CAS;
(i) B. P. Bondzic, T. Urushima, H. Ishikawa and Y. Hayashi, Org. Lett., 2010, 12, 5434 CrossRef CAS;
(j) O. Lifchits, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2010, 132, 10227 CrossRef CAS;
(k) L. Deiana, G.-L. Zhao, S. Lin, P. Dziedzic, Q. Zhang, H. Leijonmarck and A. Córdova, Adv. Synth. Catal., 2010, 352, 3201 CrossRef CAS.
- CCDC 812281 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Imidazo[1,2-a]pyridine and indolizine structural motifs and examples of biologically active imidazo[1,2-a]pyridines.](/image/article/2011/SC/c1sc00122a/c1sc00122a-f1.gif)


![X-ray structure of (R)-N-(1-(imidazo[1,2-a]pyridin-3-yl)butyl)-4-methylbenzenesulfonamide 6c.](/image/article/2011/SC/c1sc00122a/c1sc00122a-f2.gif)