High thermal and chemical stability in pyrazolate-bridged metal–organic frameworks with exposed metal sites†
Valentina
Colombo
abc,
Simona
Galli
c,
Hye Jin
Choi
ab,
Ggoch Ddeul
Han
ab,
Angelo
Maspero
c,
Giovanni
Palmisano
c,
Norberto
Masciocchi
c and
Jeffrey R.
Long
*ab
aDepartment of Chemistry, University of California, Berkeley, California 94720, U. S. A. E-mail: jrlong@berkeley.edu
bMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, U. S. A.
cDipartimento di Scienze Chimiche e Ambientali, Università dell'Insubria, via Valleggio 11, I-22100, Como, Italy
First published on 28th April 2011
Abstract
Reactions between the tritopic pyrazole-based ligand 1,3,5-tris(1H-pyrazol-4-yl)benzene (H3BTP) and transition metal acetate salts in DMF afford microporous pyrazolate-bridged metal–organic frameworks of the type M3(BTP)2·xsolvent (M = Ni (1), Cu, (2), Zn (3), Co (4)). Ab-initioX-ray powder diffraction methods were employed in determining the crystal structures of these compounds, revealing 1 and 2 to exhibit an expanded sodalite-like framework with accessible metal cation sites, while 3 and 4 possess tetragonal frameworks with hydrophobic surfaces and narrower channel diameters. Compounds 1–4 can be desolvated without loss of crystallinity by heating under dynamic vacuum, giving rise to microporous solids with BET surface areas of 1650, 1860, 930 and 1027 m2 g−1, respectively. Thermogravimetric analyses and powder X-ray diffraction measurements demonstrate the exceptional thermal and chemical stability of these frameworks. In particular, 3 is stable to heating in air up to at least 510 °C, while 1 is stable to heating in air to 430 °C, as well as to treatment with boiling aqueous solutions of pH 2 to 14 for two weeks. Unexpectedly, 2 and 3 are converted into new crystalline metal–organic frameworks upon heating in boiling water. With the combination of stability under extreme conditions, high surface area, and exposed metal sites, it is anticipated that 1 may open the way to testing metal–organic frameworks for catalytic processes that currently employ zeolites.
Introduction
A large segment of the global economy is based on the use of natural and synthetic zeolites in chemical industries as detergents, adsorbents/desiccants and heterogeneous catalysts.1 Consequently, worldwide consumption of these materials is estimated at about 4–4.5 million metric tons per year.1c,d As purely inorganic materials, zeolites are extraordinarily robust and provide moderately high surface areas, which together facilitate catalytic activity. Nevertheless, their performance can be limited by the stiffness of the framework, whose features, above all pore size and surface functionalization, are not readily modified using self-assembly approaches. Over the past decade, metal–organic frameworks have begun to emerge as possible alternatives for such applications. These materials are hybrid compounds built up from metal ions or metal-based clusters linked via various organic bridging ligands to form a three-dimensional skeleton, frequently with an extraordinarily high surface area.2 Compared to zeolites, metal–organic frameworks typically display a considerable degree of tunability, achievable by judicious selections of inorganic and organic components, or via post-synthetic modification of the surface.3 Depending upon the metal ions and organic linkers incorporated in the framework, key chemical and physical properties, such as pore size, surface area, guest binding capability, catalytic activity, can potentially be finely modulated. This has enabled researchers to generate metal–organic frameworks of interest for a variety of applications, including gas storage,4 molecular separations,5 and heterogeneous catalysis.6Although metal–organic frameworks have in rare instances displayed thermal stability up to 500 °C,7 none yet approach the robustness of zeolites, a disadvantage further worsened by problems generally related to their low chemical stability. This is particularly true for those systems based on divalent metal cations combined with organocarboxylate bridging ligands,8 which can be subject to hydrolysis and thermal decomposition in the presence of moisture.9 In this regard, it is clearly beneficial to discover new high-surface area metal–organic frameworks that are stable toward diverse environments such as air, water, acidic and basic media, and even extreme temperatures and pressures. Such advancements will extend the utility of metal–organic frameworks towards a variety of applications where zeolites have been playing a major role.
Along this line, our strategy has involved the use of polyazolate-bridging ligands,10 that can lead to frameworks with strong metal–nitrogen bonds, providing a greater chemical and thermal stability compared to their carboxylate-based counterparts. Employing polyazolate heterocycles, the strength of the resulting M–N bonds can be predicted to be closely related to the pKa values for the deprotonation of the N–H bond. Indeed, increased stability has been observed for frameworks generated from organic ligands functionalized with 1,2,3-triazole (pKa = 13.9)11 than for analogues based upon tetrazole (pKa = 4.9).11,12Imidazole, with an even higher pKa of 18.6,11 has been shown to afford frameworks of still greater thermal stability (Tdec up to 390 °C) and some chemical resistance to alkalinity and boiling solvents such as water, methanol and benzene.7a In particular, however, organic ligands functionalized with pyrazole (pKa = 19.8),11 are of interest for the synthesis of robust pyrazolate-bridged frameworks.
A number of pyrazolate-based metal–organic frameworks exhibiting exceptional stability have already been realized. For example, 1,4-bis(1H-pyrazol-4-yl)benzene (1,4-H2BDP) was found to react with salts of cobalt(II),13nickel(II) or zinc(II)7d,e to afford frameworks exhibiting good thermal stability (Tdec = 420–460 °C) and permanent porosity with Langmuir surface areas between 1600 and 2670 m2 g−1. Employing instead the bent molecule 1,3-bis(1H-pyrazol-4-yl)benzene (1,3-H2BDP) results in a double-walled zinc-based framework of even greater thermal stability (Tdec = 500 °C), which further shows chemical stability in a hot acidic solution (pH 3).7e The thermal stability of pyrazolate-based materials was again observed for the cubic frameworks Ni8L6(OH)4(H2O)2 with L = 4,4′-bis(1H-pyrazol-4-yl)biphenyl (Tdec = 420 °C) or 2,6-bis(1H-pyrazol-4-yl)pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetrone (Tdec = 410 °C).7f Furthermore, a Cu(I) framework based on 3,3′,5,5′-tetramethyl-4,4′-bipyrazolate (H2Me4bpz) was found stable up to 500 °C in nitrogen atmosphere but also in air atmosphere with a decomposition temperature of above 400 °C.7h On the whole, the thermal and chemical stability of pyrazolate-based frameworks is indeed significantly increased relative to the tetrazolate- and triazolate-bridged frameworks. We note, however, that none of these high-stability pyrazolate-based frameworks possess internal surfaces bearing open metal coordination sites.
Exposed metal cations within metal–organic frameworks have been demonstrated to lead to outstanding properties for hydrogen storage,14 gas separations,4b–e,12,15 and catalysis.6 Among the azolate-based metal–organic frameworks of this type, Mn3[(Mn4Cl)3(BTT)8]2·20MeOH (Mn-BTT, H3BTT = 1,3,5-tris(2H-tetrazol-5-yl)benzene), a rigid high-surface area framework with an expanded sodalite-like structure and exposed Mn2+ sites, exhibited a high H2 binding affinity14a and Lewis acid catalysis.6c Unfortunately, the relatively low thermal stability (Tdec ≈ 200 °C) and water-sensitivity of this tetrazolate-bridged framework limits its utility. Attempts to synthesize analogous triazolate-based structures afforded the more stable framework H3[(Cu4Cl)3(BTTri)8] (Cu-BTTri, H3BTTri = 1,3,5-tris(1H-1,2,3-triazol-5-yl)benzene).12 With improved thermal stability (Tdec = 270 °C), this compound exhibits substantial chemical resistance, retaining its porous structure in dilute HCl solution (pH 3) at room temperature or in boiling water for 3 days. Moreover, its stability in basic media enabled grafting of ethylenediamine on the open Cu2+ sites, leading to a record heat of CO2 adsorption for a metal–organic framework.
In order to achieve a still greater level of stability, approaching that of zeolites, pyrazolate-bridged analogues of this important structure type were sought. Herein, we report the synthesis of the new linker 1,3,5-tris(1H-pyrazol-4-yl)benzene (H3BTP, see Fig. 1a), and its use in generating a series of exceptionally robust metal–organic frameworks, two of which adopt the Mn-BTT structure and feature exposed metal cation sites.
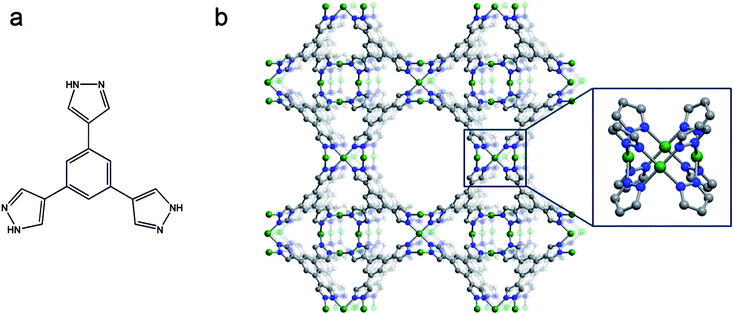 | ||
| Fig. 1 Scheme of the pyrazole-based ligand 1,3,5-tris(1H-pyrazol-4-yl)benzene, H3BTP (a) and portions of the structure of Ni3(BTP)2·3CH3OH·10H2O (1m), as determined from powder X-ray diffraction data. (b) Green, blue, and gray spheres represent Ni, N and C atoms, respectively; H atoms and solvent molecules are omitted for clarity. The inset shows the square-planar Ni4 cluster bridged by eight pyrazolate rings. The compound Cu3(BTP)2·8CH3OH·10H2O (2) is isostructural. Selected bond distances (Å) and angles (°) for the structures of 1 and 2, respectively: M–N 2.0200(4) and 2.1225(6); M⋯M 3.118(6) and 3.013(7); N–M–N 77.4(2), 102.6(2), 178.9(3) and 73.7(2), 106.0(2), 174.4(3); M–N–N 64.2(1), 116.7(1) and 67.1(1), 117.3(1). Please note that in both cases, the crystallographically independent portion of the BTP3− ligand has been modeled by means of a rigid body.19 | ||
Results and discussion
Synthesis and structure of sodalite-type Ni3(BTP)2 and Cu3(BTP)2 phases
Reaction of H3BTP with nickel(II) or copper(II) acetate in DMF at 160 °C afforded, upon washing with methanol and drying in air, Ni3(BTP)2·3DMF·5CH3OH·17H2O (1) and Cu3(BTP)2·8CH3OH·10H2O (2) as yellow and brown microcrystalline powders, respectively. Preliminary powder X-ray diffraction acquisitions showed both compounds to be isomorphous with the sodalite-like structure of Mn-BTT.14a The latter compound consists of chloride-centered [Mn4(μ4-Cl)]7+ squares linked via triangular BTT3− ligands to form a porous, three-dimensional framework in which each metal center further has a bound DMF molecule directed into the pores. Overall, the framework has an anionic charge, which is balanced by [Mn(DMF)6]2+ cations included in the pores. Despite the great similarity in size and shape between H3BTT and H3BTP, our attempts at synthesizing a Mn-BTT analogue using H3BTP and various metal chlorides were unsuccessful. Instead, the use of metal acetates in DMF was found to promote the deprotonation of the pyrazole ligand to form M–N bonds and the extended sodalite-like framework structure of 1 and 2. As assessed by X-ray powder structure analysis, 1 and 2 are isomorphous, but not isostructural with Mn-BTT. Specifically, the μ4 bridging chloride anion present in Mn-BTT, is absent in 1 and 2, as evidenced by elemental analysis and X-ray fluorescence (see Fig. S6, ESI†) and consideration on their structural features (see below).Compounds 1 and 2 crystallize (see Fig. 1) in the cubic space groupPm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, with the metal ions lying on crystallographic two-fold axes and arranged in tetranuclear cores of rigorous, crystallographically-imposed square symmetry, with M⋯M edges of 3.118(6) and 3.013(7) Å, for 1 and 2, respectively. The chloro-centered Mn4 squares in Mn-BTT showed Mn⋯Mn distances of 3.70(3) Å, in agreement with the presence of the inner μ4-Cl ion and leading to a larger accessible empty volume (as measured, e.g., by the BET specific area, vide infra).23 In 1 and 2, each M⋯M edge is bridged by pyrazolate groups from two distinct BTP3− ligands, resulting in a square-planar coordination geometry at each metal ion. Residing on a 3m crystallographic site, each BTP3− ligand employs its three pyrazolate substituents to bridge M⋯M edges of three different M4 squares. Each square is connected to eight adjacent squares, generating a rigid three-dimensional framework. Thus, the framework structure features octahedral cavities centered at [½, ½, ½], with BTP3− ligands spanning each face and M4 squares truncating each vertex to give an expanded sodalite cage unit. The sharing of squares between neighboring cage units along the three unit cell axes, results in one-dimensional channels running parallel to the cell axes. These channels have a wide diameter of nearly 10 Å (based upon van der Waals radii). Yet, only a very small entrance, possibly limiting the size and shape of adsorbable gases, allows access to the ca. 6-Å cavity within the octahedral, sodalite-like cage units. Based upon van der Waals radii, a total void volume of 66 and 69% is estimated from the structures of 1 and 2, respectively.16,17 The slight increase of unit cell and void volumes from 1 to 2 is consistent with the ionic radii of square-planar Ni2+ and Cu2+ ions (0.63 and 0.71 Å, respectively), which result in longer Cu–N bonds and a slightly expanded framework for 2. The electron density residues present in the Fourier difference maps, as resulting from the modelling of the frameworks alone, clearly indicate that: (i) both cavities and channels contain guest solvent molecules, and (ii) solvents such as DMF, CH3OH, and water can bind to open metal coordination sites. As evidenced by the isolation and characterization of different solvated forms, coordinated solvent is indeed likely to be present at an apical position, protruding into the large channels and creating a square-pyramidal coordination at each metal center.18
m, with the metal ions lying on crystallographic two-fold axes and arranged in tetranuclear cores of rigorous, crystallographically-imposed square symmetry, with M⋯M edges of 3.118(6) and 3.013(7) Å, for 1 and 2, respectively. The chloro-centered Mn4 squares in Mn-BTT showed Mn⋯Mn distances of 3.70(3) Å, in agreement with the presence of the inner μ4-Cl ion and leading to a larger accessible empty volume (as measured, e.g., by the BET specific area, vide infra).23 In 1 and 2, each M⋯M edge is bridged by pyrazolate groups from two distinct BTP3− ligands, resulting in a square-planar coordination geometry at each metal ion. Residing on a 3m crystallographic site, each BTP3− ligand employs its three pyrazolate substituents to bridge M⋯M edges of three different M4 squares. Each square is connected to eight adjacent squares, generating a rigid three-dimensional framework. Thus, the framework structure features octahedral cavities centered at [½, ½, ½], with BTP3− ligands spanning each face and M4 squares truncating each vertex to give an expanded sodalite cage unit. The sharing of squares between neighboring cage units along the three unit cell axes, results in one-dimensional channels running parallel to the cell axes. These channels have a wide diameter of nearly 10 Å (based upon van der Waals radii). Yet, only a very small entrance, possibly limiting the size and shape of adsorbable gases, allows access to the ca. 6-Å cavity within the octahedral, sodalite-like cage units. Based upon van der Waals radii, a total void volume of 66 and 69% is estimated from the structures of 1 and 2, respectively.16,17 The slight increase of unit cell and void volumes from 1 to 2 is consistent with the ionic radii of square-planar Ni2+ and Cu2+ ions (0.63 and 0.71 Å, respectively), which result in longer Cu–N bonds and a slightly expanded framework for 2. The electron density residues present in the Fourier difference maps, as resulting from the modelling of the frameworks alone, clearly indicate that: (i) both cavities and channels contain guest solvent molecules, and (ii) solvents such as DMF, CH3OH, and water can bind to open metal coordination sites. As evidenced by the isolation and characterization of different solvated forms, coordinated solvent is indeed likely to be present at an apical position, protruding into the large channels and creating a square-pyramidal coordination at each metal center.18
In examining the chemical stability of 2, some amount of the solid was refluxed in a concentrated basic (NaOH, pH 14) solution. A brown deposit isolated from the solution turned out to be a distinct new phase, Cu3(BTP)2·6H2O (2′), which could be also obtained by refluxing 2 in an acid solution (HCl, pH 3). This microcrystalline product appears, however, to be non-porous, as evidenced by a thermogravimetric analysis showing no weight loss up to decomposition (see Fig. S7, ESI†). Its powder diffraction trace could be easily indexed to a R-centered trigonal unit cell, with the likely presence of c-type glide planes. Possible space group candidates are therefore Rc and R3c, which share the same systematic extinction conditions. Indeed, a structureless Le Bail fit matched well with the observed diffraction pattern (see Table S1, ESI†). Unfortunately, compound 2′ has thus far resisted all attempts of structural resolution, although clear indications of a nearly layered disposition of the BTP3− ligands (parallel to ab) and of trinuclear Cu3 units were found. Work is in progress to assess the complete crystal structure, but the results, if any, will be postponed to a future contribution.
Synthesis and structure of tetragonal Zn3(BTP)2 and Co3(BTP)2 phases
Since the discovery of Mn-BTT, it has been established that isostructural tetrazolate-based frameworks can be synthesized with a variety of other transition-metal ions, including Cr2+, Fe2+, Co2+, Ni2+, Cu2+ and Cd2+.14,20 Similarly, we sought to obtain pyrazolate-bridged analogues of Ni3(BTP)2 and Cu3(BTP)2 incorporating other transition-metal ions with a high affinity for nitrogen-based ligands.21 After numerous attempts applying different reaction conditions, a white microcrystalline powder was obtained through addition of triethylamine to a solution of Zn(CF3SO3)2 and H3BTP in DMF. Due to the high pKa of the pyrazole rings, either base or high temperature is essential to force the reaction to proceed.22 Washing with wet methanol followed by drying in vacuo resulted in a compound of formulation Zn3(BTP)2·4CH3OH·2H2O (3). As might be expected for transition-metal ions favoring tetrahedral stereochemistry, such as zinc(II) and cobalt(II), Co3(BTP)2·8CH3OH·10H2O (4) was synthesized following the same reaction procedure.Compounds 3 and 4 crystallize in the tetragonal space groupP42/ncm. The local coordination geometry can be appreciated from the depiction at the bottom of Fig. 2, while the overall framework structure is shown at the top. The structures contain tetrahedrally coordinated metal(II) centers arranged in collinear chains running along [110] (and equivalent directions), with pyrazolate-bridged intermetallic separations of 3.748(1) and 3.654(1) Å for Co and Zn, respectively. The BTP3− ligands are bisected by a crystallographic two-fold axis and possess one pyrazolate moiety in plane with the inner arene and the other two making a dihedral angle of about 64° to the benzene core. Each chain connects to three adjacent chains to afford a three-dimensional framework. Porosity is apparent in the structures, with one-dimensional channels of slightly less than 4 Å-diameter, running parallel to c and filled with guest solvent molecules. The surfaces exposed within these channels appear to be only π-rings, thus imparting a hydrophobic character. Overall, the accessible void volume reaches 46 and 50% for the structures of 3 and 4, respectively.16 Unlike 1 and 2, these compounds do not feature metal-bound solvent molecules that could potentially be removed to generate coordinatively-unsaturated metal centers.
![Portions of the structure of Zn3(BTP)2·4CH3OH·2H2O (3), determined by powder X-ray diffraction analysis, as viewed along the c (upper) axis and [110] direction (bottom). Orange, blue and gray spheres represent Zn, N and C atoms, respectively; H atoms and solvent molecules are omitted for clarity. The compound Co3(BTP)2·8CH3OH·10H2O (4) is isostructural. Selected bond distances (Å) and angles (°) for the structures of 3 and 4, respectively: M–N 2.077(6), 2.053(6), 2.106(7) and 2.124(7), 2.035(8), 2.046(9); M⋯M 3.654(1) and 3.748(1); N–M–N 102.3(4)–127.3(4) and 97.7(4)–120.8(2); M–N–N 120.0(4), 121.3(2) 123.1(2) and 119.1(3), 119.7(3), 125.8(3). Please note that in both cases, the crystallographically independent portion of the BTP3− ligand has been modeled by means of a rigid body.19](/image/article/2011/SC/c1sc00136a/c1sc00136a-f2.gif) | ||
| Fig. 2 Portions of the structure of Zn3(BTP)2·4CH3OH·2H2O (3), determined by powder X-ray diffraction analysis, as viewed along the c (upper) axis and [110] direction (bottom). Orange, blue and gray spheres represent Zn, N and C atoms, respectively; H atoms and solvent molecules are omitted for clarity. The compound Co3(BTP)2·8CH3OH·10H2O (4) is isostructural. Selected bond distances (Å) and angles (°) for the structures of 3 and 4, respectively: M–N 2.077(6), 2.053(6), 2.106(7) and 2.124(7), 2.035(8), 2.046(9); M⋯M 3.654(1) and 3.748(1); N–M–N 102.3(4)–127.3(4) and 97.7(4)–120.8(2); M–N–N 120.0(4), 121.3(2) 123.1(2) and 119.1(3), 119.7(3), 125.8(3). Please note that in both cases, the crystallographically independent portion of the BTP3− ligand has been modeled by means of a rigid body.19 | ||
When compound 3 was heated in boiling water, a new crystalline phase Zn12[Zn2(H2O)2]6(BTP)16 (3′) was obtained, as identified by X-ray powder diffraction. The same phase was also isolated through the reaction of 3 in a concentrated basic solution (NaOH, pH 14) for 30 min. Although 3 is highly resistant to high temperatures (up to 510 °C), the solid-state transformation in basic pH conditions occurs at room temperature in a very short time, indicating that the presence of water is critical to its instability. Compound 3′ crystallizes in the cubic space groupPnn. The best structural model derived from our X-ray powder diffraction analysis was found to contain one-dimensional chains running along the three crystallographic axes. Two crystallographically distinct zinc(II) centers, referred to as Zn1 and Zn2, alternate along the chains (see Fig. 3). Site Zn1 possesses a tetrahedral stereochemistry, with coordination by four nitrogen atoms belonging to the pyrazolate moieties of four distinct BTP3− ligands. Situated at the vertices of a [Zn2(H2O)2]4+ rhombic unit, Zn2 shows a cis-ZnN2O2 tetrahedral stereochemistry, where the nitrogen atoms belong to pyrazolate groups from two distinct BTP3− ligands. Due to the orientational disorder affecting the rhombic units (which reside on a crystallographic four-fold axis), along each chain, Zn1 may be bridged, by the BTP3− ligands, either to Zn2, via a Zn–N bond, or to a water molecule, via a N⋯HO hydrogen bond (see Fig. S5, ESI†). Given the overall coordination mode of the BTP3− ligands, which employ all of their nitrogen atoms to form bonds, the chains are mutually connected to give a dense, three-dimensional framework with no voids or channels for hosting solvent. As expected in the absence of guest solvent molecules, thermogravimetric analysis shows no weight loss for the compound up to decomposition, which occurs at a rather high temperature of above 400 °C (see Fig. S8, ESI†).
![Portions of the molecular structure of Zn12[Zn2(H2O)2]6(BTP)16 (3′) analyzed by powder X-ray diffraction as viewed along a axis. Orange, red, blue and gray spheres represent Zn, O, N and C atoms, respectively; H atoms are omitted for clarity. For a description of the local disorder affecting the Zn2O2 fragment see ESI. Selected bond distances (Å) and angles (°) for the structure of 3′: Zn–N 2.07(2), 2.099(6); Zn–O 1.97(2); Zn⋯Zn 2.97(1), 3.13(5); N–Zn–N 106.7(2), 115.2(4), 140.8(2); N–Zn–O 85.3(2); O–Zn–O 82(2); Zn–N–N 101(1), 129.0(3); Zn–O–N 84(1), 129(2). Please note that in both cases, the crystallographically independent portion of the BTP3− ligand has been modeled by means of a rigid body.19](/image/article/2011/SC/c1sc00136a/c1sc00136a-f3.gif) | ||
| Fig. 3 Portions of the molecular structure of Zn12[Zn2(H2O)2]6(BTP)16 (3′) analyzed by powder X-ray diffraction as viewed along a axis. Orange, red, blue and gray spheres represent Zn, O, N and C atoms, respectively; H atoms are omitted for clarity. For a description of the local disorder affecting the Zn2O2 fragment see ESI†. Selected bond distances (Å) and angles (°) for the structure of 3′: Zn–N 2.07(2), 2.099(6); Zn–O 1.97(2); Zn⋯Zn 2.97(1), 3.13(5); N–Zn–N 106.7(2), 115.2(4), 140.8(2); N–Zn–O 85.3(2); O–Zn–O 82(2); Zn–N–N 101(1), 129.0(3); Zn–O–N 84(1), 129(2). Please note that in both cases, the crystallographically independent portion of the BTP3− ligand has been modeled by means of a rigid body.19 | ||
Gas adsorption properties
Prompted by their porous structures, we evaluated the permanent porosity of compounds 1–4 by collecting N2 adsorption isotherms at 77 K. Complete removal of coordinating solvents without collapsing the structure is not always trivial, however, due to an activation barrier which should be overcome by applying vacuum and high temperatures, often subsequent to solvent exchange using a volatile coordinating solvent such as methanol. To determine the optimal activation temperatures of the methanol-exchanged phases, the samples were heated under dynamic vacuum at gradually increasing temperatures, while N2 adsorption was repeatedly measured at each stage. From the N2 isotherm measurements, the best activation method for compounds 1 and 2 was determined to be application of dynamic vacuum at 250 °C for at least two days. With no bound solvent, 3 and 4 can be activated by heating under vacuum at the lower temperature of 160 °C for two days.The optimally desolvated materials were found to adsorb significant amounts of N2 at 77 K, displaying Type I adsorption isotherms characteristic of microporous solids (see Fig. 4). Fitting the N2 isotherms afforded BET surface areas of 1650(20), 1860(10), 930(10) and 1027(3) m2 g−1 and Langmuir surface areas of 1900(13), 2159(10), 1242(11) and 1588(40) m2 g−1 for 1, 2, 3 and 4, respectively. Perhaps owing to a smaller unit cell dimension, the surface areas of 1 and 2 are slightly lower than observed for Mn-BTT, which, thanks to the (μ4-Cl induced) inflation of the inner Mn4 core (vide supra), displayed a BET surface area of 2100 m2 g−1.14a Actually, for 3D isostructural materials sharing the same ligand and slightly different cores, also the empty volume is significantly affected by the cooperative change in size of the metallic nodes, here addressed by the M⋯M contacts in the inner M4 square (with a linear inflation, on passing from Ni4 (or Cu4) to [(μ4-Cl)Mn4], larger than 20%, see above). Similarly, a ca. 0.1 Å increase of the metal radius on passing from Cu to Pd in the sodalitic M(n-pymo)2 frameworks (n-pymo− = pyrimidin-n-olate) allowed the increase of nearly 30% of the void volume and of ca. 80% of the BET surface area.23 Notably, the increase in surface area from 1 to 2 is also consistent with their unit cell dimension and void volume, which is ultimately related to the ionic radii of the two metal ions (see above). The surface area of 3 and 4 are also consistent with that of Zn(1,3-BDP) and Co(1,3-BDP), which display much similarity in the framework connectivity and pore size.7e
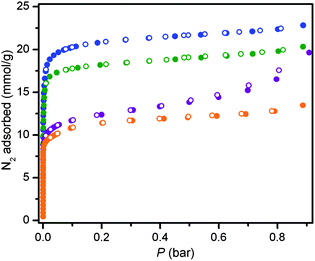 | ||
| Fig. 4 Nitrogen adsorption isotherms measured at 77 K for 1 (green), 2 (blue) 3 (orange) and 4 (purple). Filled and empty symbols represent adsorption and desorption, respectively. | ||
Thermal behavior
In order to probe the thermal stability of the new compounds, thermogravimetric analyses were performed, combined with in situ variable-temperature powder X-ray diffraction experiments. While the thermogravimetric analyses were carried out under N2 for as-synthesized compounds 1–4, complete and detailed characterization of the thermal behaviors of 1–3 were carried out in air by means of variable-temperature diffraction experiments.As depicted in Fig. 5 the thermogravimetric trace of 1 shows a weight loss of 30% between 30 and 150 °C, corresponding to the partial evolution of guest solvent (4 methanol and 16 water molecules corresponds to 30%). A gradual further weight loss of 15% occurs in the range 150–430 °C, consistent with the evolution of DMF solvent molecules coordinated to the metal sites (3 DMF molecules corresponds to 16%). Further heating prompts decomposition at 450 °C. In the TG trace of 2, a 30% weight loss occurs below 50 °C, corresponding roughly to the evolution of 5 methanol and 10 water molecules (29%). A gradual weight loss of 8% then follows up to 410 °C, consistent with the loss of 3 metal-coordinated methanol molecules, and further heating induces decomposition.
 | ||
| Fig. 5 Thermal gravimetric analysis of as-synthesized Ni3(BTP)2·3DMF·5CH3OH·17H2O (1, green), Cu3(BTP)2·8CH3OH·10H2O (2, blue), Zn3(BTP)2·4CH3OH·2H2O (3, orange) and Co3(BTP)2·8CH3OH·10H2O (4, purple). | ||
The foregoing observations are consistent with thermodiffractometric analyses (see Fig. 6 and S9, ESI†). These results confirm the high thermal stability of 1 and 2, while also showing that their crystallinity is retained to afford permanent porosity. Indeed, solvent loss does not significantly affect the crystal structures, with the powder diffraction patterns remaining largely unchanged up to 450 °C for 1 and 390 °C for 2. Notably, parametric Le Bail refinements against the data show that the two compounds respond to heat with a distinct framework flexibility. In the case of 1, the unit cell volume remains almost constant up to ca. 200 °C, while above this temperature, a modest contraction, reaching 0.5%, is observed. In comparison, the unit cell volume of 2 experiences a modest, yet continuous, decrease in the temperature range 30–350 °C, reaching 0.8% (see Fig, S10 and S11, ESI†).
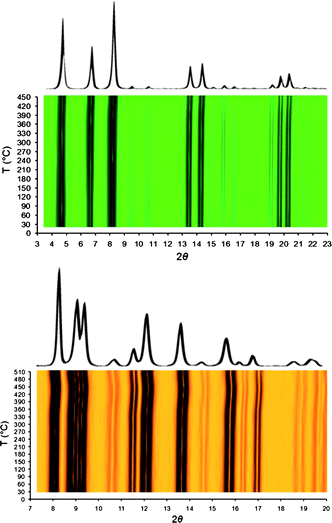 | ||
| Fig. 6 Overlaid powder X-ray diffraction patterns measured at elevated temperatures in the range 30–450 °C for 1 (upper) and 30–510 °C for 3 (lower), and their two-dimensional contour plots as a function of 2θ and temperature, both displaying their thermal stability. Notably, the diffraction patterns remained unaltered during the measurements except for minor changes in peak intensity occurring at above 400 °C. | ||
As evidenced by their thermal behavior, compounds 3 and 4 provide further examples of robust metal–organic frameworks. The thermogravimetric traces show less weight loss than expected on the basis of the pore solvent contents. For example, an 18% weight loss is expected for 3, corresponding to 4 methanol and 2 water molecules, but only a 12% loss is observed for both 3 and 4 in the temperature range 30–500 °C (see Fig. 5). This discrepancy is reasonably due to solvent evolution during weighing and transferring the sample, particularly in view of the hydrophobic nature of the pore surfaces within these compounds. After solvent removal, decomposition begins at 510 and 450 °C for 3 and 4, respectively. The remarkably high thermal stability of 3 was confirmed by diffraction measurements, which also revealed retention of the structure upon heating in air (see Fig. 6). A parametric Le Bail refinement of the data revealed this compound to be an extremely rigid material, showing a very limited volume changes upon heating. At lower temperatures, this suggests that the partial, and very limited, desolvation overcomes thermal expansion effects (see Fig. S12, ESI†). Notably, among all the members of the M3(BTP)2 family, the tetragonal zinc(II) derivative shows the greatest thermal stability. Indeed, in this regard, zinc(II) compounds have proven superior to other metal(II) analogues for all of the pyrazolate-bridged metal–organic frameworks reported so far. In the cases of M(2-pymo)2 and M(4-pymo)2 compounds, the highest tolerances to elevated temperatures have also been found for M = Zn.24
Chemical stability
The chemical resistance of 1–3 was examined by suspending samples of the compounds in boiling water, boiling aqueous HCl or HNO3 solutions at pH 2, and a boiling aqueous NaOH solution at pH 14, conditions that reflect extreme operational parameters in industry. Each sample (ca. 100 mg) was soaked in the applicable test solution, which was subsequently heated at 100 °C for two weeks. During this period of time, a portion of each sample was periodically removed, filtered, dried at room temperature and checked by X-ray powder diffraction analysis. For compound 1, after each two-week treatment, the sample was desolvated by heating at 250 °C and N2 adsorption isotherms were collected at 77 K to test retention of surface area.Remarkably, the Ni3(BTP)2 framework of 1 is stable to all of the environments tested and maintains both its crystallinity and porous nature after 14 days of uninterrupted test reactions. Powder X-ray diffraction data collected before and after each test confirm its structural chemical integrity (see Fig. 7). No change in crystallinity was observed, but only in the intensities of the peaks, which is reasonably due to the difference in solvent contents. The accessibility of the pores within the retained structure was unequivocally demonstrated by measuring the surface areas of the solid after each chemical stability test (see Table 1). Significantly, 1 retains its surface area after two weeks under all of the aforementioned extreme conditions. To our knowledge, this is the most extensive range of chemical stability yet demonstrated for a metal–organic framework. Although some frameworks are chemically resistant in a basic solution, none have been known to be stable in a pH 2 acid solution at 100 °C. Some imidazolate-based frameworks are known to be substantially retained in boiling solvents (water, methanol, benzene) for 7 days, yet only for 24 h in aqueous NaOH solution, with a poor stability in acidic solutions reported. The zirconium-based framework UIO-66,7b has been shown to display thermal stability up to 540 °C, but its chemical stability in water and common organic solvents was verified only for no longer than 24 h at room temperature. Other stability studies on tetrazolate-,10btriazolate-,12 and pyrazolate-based7e,15 frameworks have been performed but, despite their sometimes good water tolerance, the chemical stability in acidic and basic media is either inferior to 1 or not reported. Combined with its exceptional stability, the presence of exposed metal cation sites in 1, typically the preferred binding sites for adsorbates (including nonpolar species like H2), should raise its potential for a variety of applications.
 | ||
| Fig. 7 X-Ray diffraction patterns for 1 after treatment in water, acids or base for two weeks at 100 °C. | ||
| Conditions | SALangmuir/m2 g−1 |
|---|---|
| a Values were obtained from N2 adsorption measurements performed at 77 K on samples subjected to the conditions specified for two weeks and then desolvated by heating at 250 °C under dynamic vacuum. | |
| As-synthesized | 1900(13) |
| H2O | 1830(10) |
| HCl(aq) | 1791(14) |
| HNO3(aq) | 1774(11) |
| NaOH(aq) | 1925(15) |
In contrast, the copper- and zinc-based frameworks of 2 and 3, undergo transformation to non-porous crystalline solids upon extreme chemical treatment, as rather commonly observed for metal–organic frameworks. As depicted in Fig. 8, compound 2 shows a progressive phase transition in boiling water, converting to 2′. This transformation occurs upon refluxing 2 in aqueous NaOH (pH 14) or HCl (pH 3) solutions for one day. The longest resistance of 2 to pH 14 solution at room temperature was found to be one day, and it was further found to be stable for two weeks in benzene, DMF and methanol heated at reflux (see Fig. S15, ESI†). Despite its extremely high thermal stability, compound 3 displays a resistance to hot acidic media that is somewhat inferior to that of 1. While its structure is maintained upon heating at 100 °C in pH 3 aqueous HCl for 7 days, as shown in Fig. 9, it is not stable to a similar treatment at pH 2. In addition, 3 reacts in water and especially in basic solutions, transforming into the cubic phase 3′.
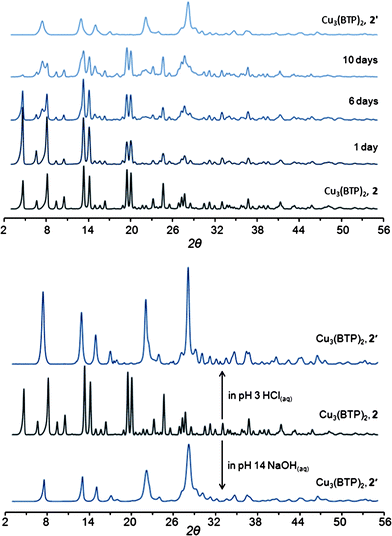 | ||
| Fig. 8 Powder X-ray diffraction patterns of 2 during treatment in water for 14 days at 100 °C (top) and transformation of 2 in 2′ after treatment in an acidic or a basic solution (bottom). | ||
 | ||
| Fig. 9 Powder X-ray diffraction patterns of 3 after treatment in water, acid or base for various durations at various temperatures. | ||
Conclusions
The foregoing results demonstrate the use of the new triangular trispyrazole molecule H3BTP in construction of microporous frameworks of the type M3(BTP)2 (M = Co, Ni, Cu, Zn) exhibiting exceptional thermal and chemical stability. In particular, Ni3(BTP)2 retains its integrity in the face of an unprecedented range of extreme conditions, including heating in air to 430 °C and treatment with boiling aqueous solutions of pH 2 to 14 for two weeks. Thus, this stability parallels, or even surpasses that of zeolites, where the presence of selectively removable Al sites makes their frameworks unstable in highly acidic and basic conditions.25 Moreover, Ni3(BTP)2 represents the first high-stability metal–organic framework with accessible metal sites lining the pore surfaces. Such a remarkable combination of properties may open the way for testing metal–organic frameworks in a variety of applications that currently employ zeolites under extreme conditions. Indeed, future efforts will focus on exploring the performance of these new high-surface area materials in various high-temperature catalytic processes, as well as on the synthesis of other pyrazolate-based metal–organic frameworks featuring exposed metal sites.Acknowledgements
This research was supported in the US by the Department of Energy under Contract No. DE-AC02-05CH11231 and in Italy by Fondazione Cariplo (Project 2007-5117). We thank Dr Leslie J. Murray, and Mr. Eric D. Bloch for helpful discussions.Notes and references
- (a) A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS; (b) T. Maesen and B. Marcus, Introduction to Zeolite Science and Practice, 2001, eds. H. Van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Elsevier, Amsterdam, ch. 1–9 Search PubMed; (c) D. H. Lauriente and Y. Inoguchi, The Chemical Economics Handbook, SRI Consulting, 2005, 599 1000 F, 14, 6 Search PubMed; (d) B. Yilmaz and U. Muller, Top. Catal., 2009, 52, 888 CrossRef CAS.
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef; (b) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (d) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- (a) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498 RSC; (b) S. M. Cohen, Chem. Sci., 2010, 1, 32 RSC; (c) Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315 RSC; (d) T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48, 3057 CrossRef CAS; (e) Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS; (f) O. K. Farha, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 10223 CrossRef CAS; (g) K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866 CrossRef CAS; (h) Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC; (i) M. J. Ingleson, J. P. Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680 RSC; (j) M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986 CrossRef CAS; (k) D. Britt, C. Lee, F. J. Uribe-Romo, H. Furukawa and O. M. Yaghi, Inorg. Chem., 2010, 49, 6387 CrossRef CAS; (l) K. Oisaki, Q. Li, H. Furukawa, A. C. Czaja and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 9262 CrossRef CAS; (m) M. Meilikow, K. Yusenko and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 9644 CrossRef CAS.
- (a) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kavazoe and Y. Mita, Nature, 2005, 436, 238 CrossRef CAS; (b) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS; (c) H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC; (d) S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 1012 CrossRef CAS; (e) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef; (f) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245 CrossRef; (g) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC , and references therein.
- (a) L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616 CrossRef CAS; (b) H. Hayashi, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS; (c) K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 6938 CrossRef CAS; (d) D. Britt, D. J. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623 CrossRef CAS; (e) D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS; (f) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (g) L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856 CrossRef CAS.
- (a) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS; (b) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS; (c) S. Horike, M. Dincă, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854 CrossRef CAS; (d) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (e) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- (a) K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS; (b) J. Hafizovic Cavka, S. Jacobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef; (c) T. Loiseau, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373 CrossRef CAS; (d) S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. Lopez-Garzon, M. Domingo-García, I. Fernandez-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664 CrossRef CAS; (e) H. J. Choi, M. Dincă, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117 RSC; (f) N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902 CrossRef CAS; (g) O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680 CrossRef CAS; (h) J.-P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907 CrossRef CAS.
- (a) S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176 CrossRef CAS; (b) M. Eddaudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS.
- (a) J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678 CrossRef CAS; (b) J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834 CrossRef CAS.
- (a) A. Cingolani, S. Galli, N. Masciocchi, L. Pandolfo, C. Pettinari and A. Sironi, J. Am. Chem. Soc., 2005, 127, 6144 CrossRef CAS; (b) M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904 CrossRef CAS; (c) A. Maspero, S. Galli, V. Colombo, G. Peli, N. Masciocchi, S. Stagni, E. Barea and J. A. R. Navarro, Inorg. Chim. Acta, 2009, 362, 4340 CrossRef CAS; (d) A. Demessence and J. R. Long, Chem.–Eur. J., 2010, 16, 5902 CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS . Note that the values given are for the non-substituted azoles, and are referenced to DMSO.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848 CrossRef CAS.
- (a) M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS; (b) M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS; (c) M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef CAS.
- E. Quartapelle-Procopio, F. Liñares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS.
- PLATON: A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C34 Search PubMed.
- Due to the extreme rigidity of the frameworks, the values quoted are representative of the void volume of the outgassed counterparts.
- Due to the heavy disorder affecting the solvent molecules in the structure, both clathrated and coordinated, their electron density has been described by a very simplified model. For further details the reader is addressed to the ESI†.
- To build the rigid model describing the ligand, the following bond distances and angles have been adopted (a) for the benzene ring: C–C = 1.39 Å; C–H = 0.95 Å; C–C–C, C–C–H = 120°; (b) for the pyrazole ring: C–C, C–N, N–N = 1.36 Å; C–H = 0.95 Å; internal ring angles = 108°; C–C–H = 126°. Cbenzene–Cpyrazole = 1.45 Å.
- (a) K. Sumida, S. Horike, S. S. Kaye, Z. R. Herm, W. L. Queen, C. M. Brown, F. Grandjean, G. J. Long, A. Dailly and J. R. Long, Chem. Sci., 2010, 1, 184 RSC; (b) K. Sumida, S. Horike, E. D. Bloch, M. L. Foo, L. J. Murray and J. R. Long, unpublished results.
- (a) N. Masciocchi, G. A. Ardizzoia, A. Maspero, G. LaMonica and A. Sironi, Inorg. Chem., 1999, 38, 3657 CrossRef CAS; (b) A. P. Sadimenko and S. S. Basson, Coord. Chem. Rev., 1996, 147, 247 CrossRef CAS.
- (a) W. Evans, J. Chem. Educ., 2004, 81, 1191 CrossRef CAS; (b) J. G. Vos and W. L. Groeneveld, Transition Met. Chem., 1979, 4, 137 CrossRef CAS.
- J. A. R. Navarro, E. Barea, J. M. Salas, N. Masciocchi, S. Galli, A. Sironi, C. O. Ania and J. B. Parra, Inorg. Chem., 2006, 45, 2397 CrossRef CAS.
- (a) N. Masciocchi, G. A. Ardizzoia, G. LaMonica, A. Maspero and A. Sironi, Eur. J. Inorg. Chem., 2000, 2507 CrossRef CAS; (b) E. Barea, J. A. R. Navarro, J. M. Salas, N. Masciocchi, S. Galli and A. Sironi, Inorg. Chem., 2004, 43, 473 CrossRef CAS.
- (a) A. Cizmek, L. Komunjer, B. Subotic, M. Siroki and S. Roncevic, Zeolites, 1991, 11, 258 CAS; (b) A. Cizmek, L. Komunjer, B. Subotic, M. Siroki and S. Roncevic, Zeolites, 1991, 11, 810 CAS; (c) A. Cizmek, L. Komunjer, B. Subotic, M. Siroki and S. Roncevic, Zeolites, 1992, 12, 190 CrossRef CAS; (d) A. Petushkov, J. Freeman and S. C. Larsen, Langmuir, 2010, 26, 6695 CrossRef CAS; (e) R. L. Hartman and H. S. Fogler, Langmuir, 2007, 23, 5477 CrossRef CAS; (f) K. J. Murata, J. Am. Mineral., 1943, 28, 545 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic files (CIF), detailed experimental procedures, crystal data and plots showing additional data (PDF). See DOI: 10.1039/c1sc00136a |
| This journal is © The Royal Society of Chemistry 2011 |