Mild and selective boronic acid catalyzed 1,3-transposition of allylic alcohols and Meyer–Schuster rearrangement of propargylic alcohols†
Hongchao
Zheng
,
Michal
Lejkowski
and
Dennis G.
Hall
*
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: dennis.hall@ualberta.ca
First published on 21st April 2011
Abstract
Boronic acid catalysis (BAC) was applied to the 1,3-transposition of allylic alcohols and the related Meyer–Schuster rearrangement of propargylic alcohols using highly electron deficient polyfluoroarylboronic acids as catalysts under mild metal-free conditions. A wide range of synthetically useful products are formed in E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z selectivities superior to that of metal-catalyzed methods. A mechanism is proposed involving partial or full ionization into an allylic (or propargylic) carbocation, and additional possibilities for multicatalytic tandem reactions are exemplified.
:Z selectivities superior to that of metal-catalyzed methods. A mechanism is proposed involving partial or full ionization into an allylic (or propargylic) carbocation, and additional possibilities for multicatalytic tandem reactions are exemplified.
New ways to catalyze organic reactions are being sought continually in order to expand the substrate scope and the selectivity of existing transformations, or to enable new reactions for the production of organic compounds of interest in medicine and materials science.1 In this regard, boronic acid catalysis (BAC) is emerging as a mild and effective strategy for the direct activation of alcohols and carboxylic acids.2 Important reactions such as direct esterification and amidation,3imine hydrolysis,4epoxide opening,5Biginelli reaction,6 Diels–Alder7 and dipolar cycloadditions,8aldol condensation,9ene reaction,10 and Friedel–Crafts alkylations11 have all been performed recently under BAC. Faster reactions, milder conditions, and increased selectivity are some of the benefits provided by BAC. The 1,3-transposition of allylic alcohols12 and the related Meyer–Schuster rearrangement of propargylic alcohols13 are synthetically useful processes that can be catalyzed in various manners. Several methods, however, require stoichiometric activation of the hydroxyl group,14 or the use of transition metals15 or strong protic acid catalysts16 often under harsh conditions such as high temperature. Although rhenium(VII) oxo complexes are generally very effective catalysts,12,17 they need to be stored and used under strictly anhydrous conditions. Based on recent reports from McCubbin and co-workers describing mild Friedel–Crafts alkylations by activation of allylic/benzylic alcohols with air-stable boronic acids,11 we reasoned that allylic alcohols could rearrange selectively in the absence of an external nucleophile.
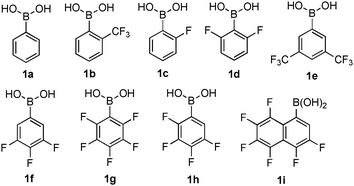
In the first round of optimization, a large number of arylboronic acids were evaluated for their ability to accelerate the allylic rearrangement of 1-phenylprop-2-en-1-ol (2a) as a model alcohol (see ESI†). Further optimization focused on the most active class of electron-poor arylboronic acids (Table 1). Just like the recently reported Friedel–Crafts alkylations,11 highly electron-deficient polyfluorinated arylboronic acids are preferable. Thus, at the onset pentafluorophenylboronic acid (1g) stood out as a promising catalyst (entry 7). By taking into account our previous observations in the catalysis of direct amidations with ortho-halogenated arylboronic acids,3f we reasoned that the removal of one of the orthofluoride substituents could provide steric relief and accelerate the rearrangement despite the attenuation of electronic effects. In the event, 2,3,4,5-tetrafluorophenylboronic acid (1h) was found to be significantly superior to 1g (entry 7 vs. 8, Table 1), which supports the above steric hypothesis. Based on these steric and electronic factors, hexafluoronaphthalene catalyst 1i was designed and showed to be even superior to 1h (entry 9).

|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Loading (mol%) | Solvent b | T/°C | t/h | Yieldc (%) |
| a Reaction conditions: to a solution of 0.4 mmol of 1-phenylprop-2-en-1-ol (2a) in the indicated solvent (1 mL) was added the boronic acid catalyst (0.04–0.2 mmol). The solution was stirred at room temperature for 48 h. b Dry, column-purified solvent was employed. c Isolated yields of product purified by column chromatography on silica gel. d 4A molecular sieves (0.5 g) were added. e One equivalent of water was added. | ||||||
| 1 | 1a | 20 | Toluene | rt | 48 | 0 |
| 2 | 1b | 20 | Toluene | rt | 48 | 0 |
| 3 | 1c | 20 | Toluene | rt | 48 | 0 |
| 4 | 1d | 20 | Toluene | rt | 48 | 0 |
| 5 | 1e | 20 | Toluene | rt | 48 | <5 |
| 6 | 1f | 20 | Toluene | rt | 48 | <5 |
| 7 | 1g | 20 | Toluene | rt | 48 | 10 |
| 8 | 1h | 20 | Toluene | rt | 48 | 36 |
| 9 | 1i | 20 | Toluene | rt | 48 | 72 |
| 10 | 1h | 20 | CH2Cl2 | rt | 48 | 26 |
| 11 | 1h | 20 | DMF | rt | 48 | 17 |
| 12 | 1h | 20 | THF | rt | 48 | 7 |
| 13 | 1h | 20 | MeOH | rt | 48 | 0 |
| 14 | 1h | 50 | Toluene | rt | 48 | 35 |
| 15 | 1h | 10 | Toluene | rt | 48 | 14 |
| 16 | 1h | 20 | Toluene d | rt | 48 | 18 |
| 17 | 1h | 20 | Toluene e | rt | 48 | 5 |
Further optimization of solvent and catalyst loading confirmed that the use of 20 mol% 1h or 1i in toluene provided the best reaction conditions. A number of simple additives were screened but provided no rate acceleration.18 Furthermore, the use of molecular sieves was detrimental, while excess water suppressed the reaction (entries 16 and 17, Table 1). These observations suggest that a small quantity of water is required for the catalytic turnover, but that a larger excess interferes with formation of reactive intermediates (vide infra).
The scope of substrates was explored using the optimal reaction conditions (20 mol% catalyst 1h or 1i in toluene). While commercially available catalyst 1h was suitable with many substrates, catalyst 1i was employed with the more difficult ones. Secondary allylic alcohols substituted with an aryl or heteroaryl group provided good to excellent yields of products (entries 1-4, Table 2). Substrate 2b with an electron-releasing substituent reacted more readily (entry 2), while a substrate (2c) with a weakly electron-withdrawing chloride substituent required a higher temperature (entry 3). It is noteworthy that substrate 2b with an acid-sensitive phenolic silyl group tolerated these very mild conditions. Secondary alcohol substrates substituted with an aliphatic side chain were unreactive under the same conditions (data not shown). All tertiary alcohols, however, were reactive and provided the desired allylic alcohols with a trisubstituted alkene (entries 5–12). All examples of entries 6–9 were highly E:Z selective and significantly more so than the corresponding rhenium oxo catalyzed reactions.19 These rearrangements occur readily at room temperature except for the fully aliphatic substrates 2h and 2i, which required an elevated temperature (entries 8 and 9). The isomerization of linalool into geraniol (entry 9), an industrial process, proceeded with a selectivity superior to that of vanadium and rhenium oxo catalysts.19 Moreover, rhenium oxo catalysis failed on functionalized substrates such as 2k.19 Substitution on the alkene was well tolerated (entries 13 and 15) provided the substituent is not a strongly electron-withdrawing group such as a carboxyester (entry 14). A cyclic tertiary alcohol, 2p, provided the expected product 3p in a good yield (entry 16).
| Entry | Substrate | Catalyst | T/°C | t/h | Product |
E:Zb |
Yieldc (%) |
|---|---|---|---|---|---|---|---|
|
a
Reaction conditions: to a solution of allylic alcohol substrate (0.4 mmol) in toluene (1 mL) was added the boronic acid catalyst (0.08 mmol). The solution was stirred at the indicated temperature for the indicated period of time.
b The E:Z ratio was measured by 1H NMR of the crude reaction product.
c Isolated yields of product purified by column chromatography on silica gel. In most cases, other materials are minor amounts of leftover substrate and elimination product.
d The yield in parentheses is for the reaction performed at room temperature for 48 h.
e The yield in parentheses is for the reaction employing 1h as the catalyst at room temperature for 48 h.
f The yield in parentheses is for the reaction performed at room temperature for 48 h.
|
|||||||

|

|
||||||
| 1 | 2a (R = H) | 1i | 50 | 48 | 3a (R = H) | >20:1 |
93 (72)d |
| 2 | 2b (R = OTIPS) | 1i | rt | 48 | 3b (R = OTIPS) | >20:1 |
75 (68)e |
| 3 | 2c (R = Cl) | 1i | 50 | 48 | 3c (R = Cl) | >20:1 |
67 |
| 4 |

|
1i | rt | 48 |

|
>20:1 |
75 |
|

|
||||||
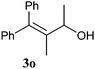
| 5 | 2e (R = Ph) | 1h | rt | 24 | 3e (R = Ph) | — | 80 |
| 6 | 2f (R = Et) | 1h | rt | 24 | 3f (R = Et) | 8:1 |
72 |
| 7 |
|
1h | rt | 48 |

|
1:20 |
76 |
| 8 |

|
1i | 80 | 48 |

|
20:1 |
77 |
| 9 |

|
1i | 80 | 48 |

|
6:1 |
62 |

|
|
||||||
| 10 | 2j (PG = Fmoc) | 1i | 50 | 12 | 3j (PG = Fmoc) | — | 81 |
| 11 | 2k (PG = t-Boc) | 1i | 50 | 12 | 3k (PG = t-Boc) | — | 72 |
| 12 | 2l (PG = Cbz) | 1i | 50 | 12 | 3l (PG = Cbz) | — | 74 |
| 13 |

|
1h | rt | 4 |

|
>20:1 |
73 |
| 14 |

|
1h | 80 | 48 |

|
5:1 |
20 (0)f |
| 15 |

|
1h | rt | 48 |
|
— | 71 |
| 16 |

|
1i | rt | 14 |

|
— | 75 |
The corresponding transposition of propargylic alcohols, known as the Meyer–Schuster rearrangement, is a useful two-step alternative process to phosphorus-based olefination methods.13 The facile addition of acetylide anions onto aldehydes and ketones provides the requisite propargylic alcohols, and BAC was shown to be very effective on those substrates. While a secondary alcohol substrate with a terminal alkyne required catalyst 1i (entry 1, Table 3), a tertiary alcohol rearranged efficiently in short time with 1h at room temperature to give the enal product in high yield (entry 2). A tertiary propargylic alcohol with a disubstituted alkyne also reacted readily to provide the enone product in high yield (entry 3). Finally, 1-ethoxy alkynyl carbinols15b rearrange readily, as shown with 4d, which gave the α,β-unsaturated ester 5d in a good yield with high E:Z selectivity when using thiophenol as an additive for in situisomerization (entry 4). Contrary to the analogous allylic alcohols, a secondary propargylic alcohol with a hindered aliphatic side chain successfully rearranged to give α,β-unsaturated ester 5e in high yield and high E:Z selectivity (entry 5). Remarkably, unsaturated thioesters and amides were also prepared under similar conditions (entries 7 and 8).
| Entry | Substrate | Catalyst | T/°C | t/h | Product |
E:Zb |
Yieldc (%) |
|---|---|---|---|---|---|---|---|
|
a
Reaction conditions: to a solution of propargylic alcohol substrate (0.4 mmol) in toluene (1 mL) was added the boronic acid catalyst (0.08 mmol). The solution was stirred at the indicated temperature for the indicated period of time.
b The E:Z ratio was measured by 1H NMR of the crude reaction product.
c Isolated yields of product purified by column chromatography on silica gel. In most cases, other materials are minor amounts of leftover substrate.
d The yield in parentheses is for the reaction performed with 1h as the catalyst at 80 °C for 48 h.
e The initial E:Z ratio is 4:3, improved to >20:1 using in situPhSH promoted isomerization.
f The initial E:Z ratio is 3:1, improved to >20:1 using in situPhSH promoted isomerization.
|
|||||||
| 1 |

|
1i | 50 | 6 |

|
>20:1 |
75 (0)d |
| 2 |

|
1h | rt | 0.3 |

|
— | 87 |
| 3 |

|
1h | rt | 0.3 |

|
— | 90 |
| 4 |

|
1h | rt | 2 |

|
>20:1e |
80 |
| 5 |

|
1i | 50 | 6 |

|
>20:1 |
78 |
| 6 |

|
1i | 50 | 1 |

|
— | 89 |
| 7 |

|
1i | rt | 0.5 |

|
— | 88 |
| 8 |

|
1i | rt | 24 |

|
>20:1f |
80 |
As demonstrated with this broad panel of substrates, it is noteworthy that contrary to traditional rhenium and gold catalysts, BAC is effective with both allylic and propargylic alcohols.19 Moreover, it tolerates both acid and base sensitive groups. The boronic acid catalyst is robust under the reaction conditions and is still fully effective even after 24 h in the reaction mixture.20
The scope of substrates, reaction times, and product yields of these 1,3-transpositions of allylic and propargylic alcohols are consistent with a pseudo SN1′ mechanism involving partial or full ionization into an allylic (or propargylic) carbocation.21 Thus, substrates bearing multiple alkyl and aryl substituents on the 3-carbon framework favored a faster transposition while substrates with fewer substituents such as 2a, or others such as 2c or 2n with electron-withdrawing groups were less favorable. In this context, a highly electron-poor boronic acid catalyst is required in order to help ionize the hydroxyl C–O bond. These assumptions and our observations about the requirement for a small amount of water are depicted in the proposed catalytic cycle of Scheme 1.22 In this cycle, the water released in the formation of hemiboronic ester 6 eventually serves in the hydrolytic release of the catalyst from the transposed intermediate 7.
 | ||
| Scheme 1 Proposed catalytic cycle for the 1,3-transposition of allylic alcohols catalyzed by boronic acid 1h or 1i. | ||
The stereochemistry of the boronic acid catalyzed transpositions of allylic alcohols can provide important mechanistic insight. When treated with catalyst 1h under the same conditions, optically enriched substrates 8 and 10 led to the respective products 9 and 11 (Scheme 2). The absolute configuration of products is consistent with a cyclic chairlike transition state similar to that proposed with the analogous rhenium-catalyzed reactions.17e,f,i However, the observation that 10 preserved much of its optical purity compared to 8 hints at a substrate-dependent degree of ionization and concertedness for the transposition. In the case of 8, recovered substrate from an incomplete reaction kept its stereochemical integrity, and product 9 did not show any decrease of optical purity upon prolonged exposure to the reaction conditions (see ESI†). These results suggest that the formation of hemiboronic acid intermediate 6 (Scheme 1) is likely irreversible, and that epimerization occurs on the latter intermediate and does so more readily on substrates capable of greater stabilization of an allylic carbocation. The 1,3-transpositions of 18O labeled substrates 2e and 4b were performed to further investigate the mechanism.23 As shown in Scheme 3, the isomerization could proceed through two possible pathways; a SN1′ pathway via a carbocation (open transition state) or a SN2′ pathway via a cyclic chairlike transition state. If the reaction proceeds through the SN1′ pathway, the three OH groups in the tetrahedral boronate counteranion would have an equal chance to attack the intermediate carbocation. Thus, when a stoichiometric amount of 1h is employed, it is statistically expected that one third of the labeled oxygen atom would transfer from the starting material to the final product. If the reaction proceeds through a concerted cyclic chairlike transition state, very little or close to none of the labeled oxygen atom would be expected to transfer to the final product. The experimental data for isomerization of 2e showed that 10.1% of the labeled oxygen atom was transferred to the final product, which is consistent with a high degree of concertedness in the transposition of allylic alcohols (cf.TS, Scheme 1). The experimental data for the 1,3-transposition of 4b showed that 33.2% 18O atom was transferred to the final product, which clearly supports the involvement of a non-concerted process with propargylic alcohols, presumably due to geometrical constraints.23
 | ||
| Scheme 2 Stereochemical study of the 1,3-transposition of allylic alcohols. | ||
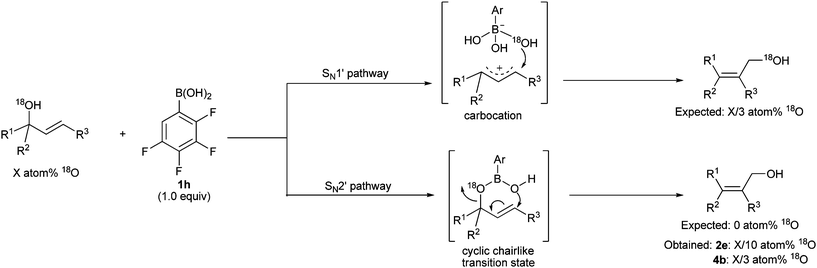 | ||
| Scheme 3 Mechanistic investigation of the 1,3-transposition of allylic and propargylic alcohols using 18O labeling experiments. | ||
Although the 1,3-transpositions of allylic alcohols are potentially reversible, they are driven by the formation of thermodynamically favorable alkene products with a higher degree of substitution or with extended conjugation. Thus, mixtures of products will be observed only in specific cases such as 12, which rearranges to give a similarly stable product, 13 (eqn (1)). Aliphatic alcohol products such as 9, however, can react further and undergo a dehydrative elimination to give substituted butadienes like 14 when treated with catalyst 1h for an extended time (Scheme 4).24
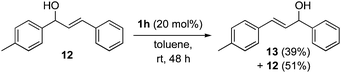
| (1) |
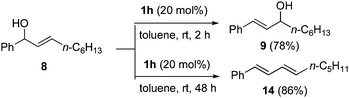 | ||
| Scheme 4 Selective time-controlled elimination to form substituted 1,3-butadienes. | ||
This additional process expands the realm of possibilities in the design of multicatalytic tandem reactions using the BAC concept. For example, allylic alcohol 2m can be subjected to a one-pot sequential 1,3-transposition/elimination/[4 + 2] cycloaddition/amidation using three different boronic acid catalysts previously identified to be optimal for these respective reactions (Scheme 5).7,3f
 | ||
| Scheme 5 Multicatalytic tandem reaction process. | ||
Conclusions
In summary, we have reported the application of boronic acid catalysis (BAC) to the 1,3-transposition of allylic alcohols and the corresponding Meyer–Schuster rearrangement of propargylic alcohols. BAC provides mild reaction conditions using air-stable catalysts that are effective with a synthetically useful range of substrates, and affords higher E:Z selectivity compared to transition-metal catalysts. The additional feasibility for dehydrative elimination leads to attractive possibilities in the design of multicatalytic tandem processes using the growing number of reactions susceptible to BAC.
Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Steacie Memorial Fellowship to D. G. H.), and the University of Alberta. H. Z. thanks the Alberta Ingenuity Foundation for a Graduate Scholarship. The authors thank Blake Lazurko for help with HPLC analyses and Michal Nicpon for the preparation of starting materials.Notes and references
- Concepts of Modern Catalysis and Kinetics: 2nd and Completely Revised and Enlarged Edition, ed. I. Chorkendorff and J. W. Niemantsverdriet, Wiley-VCH, Weinheim, 2007 Search PubMed.
- I. Georgiou, G. Ilyashenko and A. Whiting, Acc. Chem. Res., 2009, 42, 756–768 CrossRef CAS.
- (a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS; (b) P. Wipf and X. Wang, J. Comb. Chem., 2002, 4, 656–660 CrossRef CAS; (c) K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS; (d) T. Maki, K. Ishihara and H. Yamamoto, Tetrahedron, 2007, 63, 8645–8657 CrossRef CAS; (e) S. M. Levonis, L. F. Bornaghi and T. A. Houston, Aust. J. Chem., 2007, 60, 821–823 CrossRef CAS; (f) R. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS; (g) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124–134 RSC; (h) M. Tommaso, Angew. Chem., Int. Ed., 2010, 49, 6840–6843 CrossRef CAS; (i) A. Sakakura, T. Ohkubo, R. Tamashita, M. Akakura and K. Ishihara, Org. Lett., 2011, 13, 892–895 CrossRef CAS.
- G. Rao and M. Philipp, J. Org. Chem., 1991, 56, 1505–1512 CrossRef CAS.
- X.-D. Hu, C.-A. Fan, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2004, 43, 1702–1705 CrossRef CAS.
- A. Debache, B. Boumoud, M. Amimour, A. Belfaitah, S. Rhouati and B. Carboni, Tetrahedron Lett., 2006, 47, 5697–5699 CrossRef CAS.
- H. Zheng and D. G. Hall, Tetrahedron Lett., 2010, 51, 3561–3564 CrossRef CAS.
- H. Zheng, R. McDonald and D. G. Hall, Chem.–Eur. J., 2010, 16, 5454–5460 CAS.
- K. Arnold, A. S. Batsanov, B. Davies, C. Grosjean, T. Schuetz, A. Whiting and K. Zawatzky, Chem. Commun., 2008, 3879–3881 RSC.
- M. Li, T. Yang and D. J. Dixon, Chem. Commun., 2010, 46, 2191–2193 RSC.
- (a) J. A. McCubbin, H. Hosseini and O. V. Krokhin, J. Org. Chem., 2010, 75, 959–962 CrossRef CAS; (b) J. A. McCubbin and O. V. Krokhin, Tetrahedron Lett., 2010, 51, 2447–2449 CrossRef CAS.
- For reviews, see: (a) S. Bellemin-Laponnaz and J.-P. Le Ny, C. R. Chim., 2002, 5, 217–224 CrossRef CAS; (b) V. Cadierno, P. Crochet and J. Gimeno, Synlett, 2008, 1105–1124 CAS.
- For reviews, see: (a) S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS; (b) D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149–4158 RSC; (c) V. Cadierno, P. Crochet, D. E. Garcia-Garrido and J. Gimeno, Dalton Trans., 2010, 39, 4015–4031 RSC. For recent references on the Meyer–Schuster rearrangement of propargylic alcohols, see: (d) D. A. Engel and G. B. Dudley, Org. Lett., 2006, 8, 4027–4029 CrossRef CAS; (e) K. Tanaka, T. Shoji and M. Hirano, Eur. J. Org. Chem., 2007, 2687–2699 CrossRef CAS; (f) S. I. Lee, J. Y. Baek, S. H. Sim and Y. K. Chung, Synthesis, 2007, 2107–2114 CAS; (g) M. Egi, Y. Yamaguchi, N. Fujiwara and S. Akai, Org. Lett., 2008, 10, 1867–1870 CrossRef CAS; (h) D. A. Engel, S. S. Lopez and G. B. Dudley, Tetrahedron, 2008, 64, 6988–6996 CrossRef CAS; (i) L. Ye and L. Zhang, Org. Lett., 2009, 11, 3646–3649 CrossRef CAS; (j) R. S. Ramon, N. Marion and S. P. Nolan, Tetrahedron, 2009, 65, 1767–1773 CrossRef CAS; (k) M. Georgy, V. Boucard, O. Debleds, C. Dal-Zotto and J. M. Campagne, Tetrahedron, 2009, 65, 1758–1766 CrossRef CAS; (l) V. Cadierno, J. Francos and J. Gimeno, Tetrahedron Lett., 2009, 50, 4773–4776 CrossRef CAS; (m) M. Stefanoni, M. Luparia, A. Porta, G. Zanoni and G. Vidari, Chem.–Eur. J., 2009, 15, 3940–3944 CrossRef CAS; (n) R. S. Ramon, S. Gaillard, A. M. Z. Slawin, A. Porta, A. D'Alfonso, G. Zanoni and S. P. Nolan, Organometallics, 2010, 29, 3665–3668 CrossRef CAS; (o) M. N. Pennell, M. G. Unthank, P. Turner and T. D. Sheppard, J. Org. Chem., 2011, 76, 1479–1482 CrossRef CAS; (p) Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS.
- For recent references on the 1,3-transposition of allylic esters, silyl ethers, orthoesters, and others, see: (a) E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2006, 128, 8142–8143 CrossRef CAS; (b) R. E. Conrow, Org. Lett., 2006, 8, 2441–2443 CrossRef CAS; (c) S. Shekhar, B. Trantow, A. Leitner and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 11770–11771 CrossRef CAS; (d) G. Zanoni, A. D'Alfonso, A. Porta, L. Feliciani and S. P. Nolan, Tetrahedron, 2010, 66, 7472–7478 CrossRef CAS; (e) A. Serra-Muns, A. Guerinot, S. Reymond and J. Cossy, Chem. Commun., 2010, 46, 4178–4180 RSC.
- (a) V. Cadierno, S. E. Garcia-Garrido and J. Gimeno, Adv. Synth. Catal., 2006, 348, 101–110 CrossRef CAS; (b) S. S. Lopez, D. A. Engel and G. B. Dudley, Synlett, 2007, 949–953 CAS; (c) B. M. Trost and R. C. Livingston, J. Am. Chem. Soc., 2008, 130, 11970–11978 CrossRef CAS; (d) M. Egi, Y. Yamaguchi, N. Fujiwara and S. Akai, Org. Lett., 2008, 10, 1867–1870 CrossRef CAS; (e) Y. S. Vikhe, S. M. Hande, N. Kawai and J. Uenishi, J. Org. Chem., 2009, 74, 5174–5180 CrossRef CAS; (f) P. C. Ravikumar, L. Yao and F. F. Fleming, J. Org. Chem., 2009, 74, 7294–7299 CrossRef CAS.
- R. R. Leleti, B. Hu, M. Prashad and O. Repic, Tetrahedron Lett., 2007, 48, 8505–8507 CrossRef CAS.
- For selected and recent examples with allylic alcohols: (a) S. Bellemin-Laponnaz, H. Gisie, J. P. Le Ny and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1997, 36, 976–978 CrossRef CAS; (b) J. Jacob, J. H. Espenson, J. H. Jensen and M. S. Gordon, Organometallics, 1998, 17, 1835–1840 CrossRef CAS; (c) S. Bellemin-Laponnaz, J. P. Le Ny and J. A. Osborn, Tetrahedron Lett., 2000, 41, 1549–1552 CrossRef CAS; (d) F. R. Fronczek, R. L. Luck and G. Wang, Inorg. Chem. Commun., 2002, 5, 384–387 CrossRef CAS; (e) G. Wang, A. Jimtaisong and R. L. Luck, Organometallics, 2004, 23, 4522–4525 CrossRef CAS; (f) C. Morrill and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 2842–2843 CrossRef CAS; (g) G. Wang, A. Jimtaisong and R. L. Luck, Inorg. Chim. Acta, 2005, 358, 933–940 CrossRef CAS; (h) S. Akai, K. Tanimoto, Y. Kanao, M. Egi, T. Yamamoto and Y. Kita, Angew. Chem., Int. Ed., 2006, 45, 2592–2595 CrossRef CAS; (i) C. Morrill, G. L. Beutner and R. H. Grubbs, J. Org. Chem., 2006, 71, 7813–7825 CrossRef CAS; (j) M. Uyanik, R. Fukatsu and K. Ishihara, Org. Lett., 2009, 11, 3470–3483 CrossRef CAS; (k) A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962–5963 CrossRef CAS.
- Additives tried without success: TsOH (20 mol%) and ZrCl4 (20 mol%).
- Use of optimal O3ReOSiPh3 catalyzed conditions provided the following outcome (yield (%) (E:Z)): entry 8: 71 (5:1); entry 9: 63 (1:3.5); entry 10: 85 (5:1); entry 11: 81 (3:1). Use of AuCl3 catalyzed conditions for entry 25: 92%, 10:1 E:Z. Au catalysis (AuCl3) failed with allylic alcohol 1a. Re catalysis failed with allylic alcohol 2k, and propargylic alcohols 4a and 4c. See ESI† for more details.
- After completion of a reaction with substrate 2e (24 h) under optimized reaction conditions (Table 2), an additional equal amount of starting material 2e was added to the reaction mixture. Upon stirring for another 24 h, the reaction gave the desired product 3e in the same yield (80%). See ESI† for more details.
- For a related, original discussion of similar mechanisms using metal-oxo catalysts, see: P. Chabardes, E. Kuntz and J. Varagnat, Tetrahedron, 1977, 33, 1775–1783 Search PubMed.
- Protic acid catalysis can be ruled out based on the weak acidity of boronic acids and an unsuccessful attempt to catalyze the reaction of 2a with TsOH·H2O under the same conditions as Table 2, entry 1.
- See ESI† for more details.
- For similar examples, see: (a) S. Park and D. Lee, Synthesis, 2007, 2313–2316 CAS; (b) K. Narasaka, H. Kusama and Y. Hayashi, Tetrahedron, 1992, 48, 2059–2068 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and NMR spectral reproductions for new compounds, including the preparation of catalyst 1i. See DOI: 10.1039/c1sc00140j |
| This journal is © The Royal Society of Chemistry 2011 |