Terminal hafnium phosphinidene complexes and phosphinidene ligand exchange†
Rory
Waterman‡
and
T. Don
Tilley
*
Department of Chemistry, University of California, Berkeley, California 94720-1460, USA. E-mail: tdtilley@berkleey.edu; Fax: +01 510 642-8940; Tel: +01 510 642-8939
First published on 28th April 2011
Abstract
Thermolysis of the structurally characterized hafnium phosphide complex, CpCp*HfMe(PHPh) (2), resulted in formation of the triphosphanato compound CpCp*Hf(P3Ph3) (3). Trapping reactions with PMe3 gave evidence for an intermediate phosphinidene complex, which was corroborated by synthesis of the related 2,6-dimesitylphenyl derivative CpCp*(Me3P)Hf![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(dmp) (8). The hafnocene phenylphosphinidene intermediate can also be intercepted by a [2 + 2]-cycloaddition reaction with 2-butyne. However, reaction of 2 with either xylyl isocyanide or benzophenone gives insertion into the hafnium methyl bond. Under thermolytic conditions, metal dichloro complexes can efficiently intercept a phosphinidene fragment from 2 in a unique phosphinidene ligand exchange reaction. Thus, complex 2 reacts with (dippe)PtCl2 (11, dippe = 1,2-bis(diisopropylphosphino)ethane) and [N(Np)Ar]3TaCl2 (14, Np = neopentyl, Ar = 3,5-Me2C6H3) to afford phosphinidene complexes [(dippe)Pt(μ-PPh)]2 (12) and [N(Np)Ar]3TaPPh (15), respectively, in good isolated yields. A derivative of 12, [(dippe)Pt]2(μ-PPh) (13), was structurally characterized.
P(dmp) (8). The hafnocene phenylphosphinidene intermediate can also be intercepted by a [2 + 2]-cycloaddition reaction with 2-butyne. However, reaction of 2 with either xylyl isocyanide or benzophenone gives insertion into the hafnium methyl bond. Under thermolytic conditions, metal dichloro complexes can efficiently intercept a phosphinidene fragment from 2 in a unique phosphinidene ligand exchange reaction. Thus, complex 2 reacts with (dippe)PtCl2 (11, dippe = 1,2-bis(diisopropylphosphino)ethane) and [N(Np)Ar]3TaCl2 (14, Np = neopentyl, Ar = 3,5-Me2C6H3) to afford phosphinidene complexes [(dippe)Pt(μ-PPh)]2 (12) and [N(Np)Ar]3TaPPh (15), respectively, in good isolated yields. A derivative of 12, [(dippe)Pt]2(μ-PPh) (13), was structurally characterized.
Introduction
Complexes bearing terminal phosphinidene ligands, LnMPR, have received considerable attention for their unique stoichiometric and catalytic reactivity.1 An interesting development in this field was the report by Mindiola and Protasiewicz of a phospha-Wittig reagent that allows phosphinidene-group transfers to give terminal phosphinidene complexes of vanadium and zirconium.2 Additionally, Mindiola and co-workers have recently reported a scandium–lithium complex with a bridging phosphinidene ligand, which behaves as a phosphinidene-transfer reagent in formation of several compounds including a zirconium phosphinidene complex.3 Earlier reports of the use of metal complexes in phosphinidene transfers to organic substrates have produced (for example) phosphirane, phosphirene and phospholene compounds.1d,1g,4
Group 4 metal complexes have been at the forefront of terminal phosphinidene chemistry. The first examples of isolated group 4 phosphinidene complexes, of the general form Cp2(L)ZrPR (L = neutral donor), were reported by Stephan and co-workers.1d,5 Studies on complexes of this type have provided considerable information regarding the reactivity of so-called “nucleophilic” phosphinidene species.1b,6 Such zirconocene complexes may also be important in the catalytic dehydrocoupling of phosphines.7 Mindiola has shown that a terminal titanium phosphinidene derivative may serve as a catalyst for the hydrophosphination of alkynes.8
Transition metal–phosphorus complexes are therefore of interest as phosphinidene transfer reagents and catalysts, and research along these lines has pointed to phosphinidene complexes, LnMPR, as key intermediates. However, another possibility for the development of phosphinidene-transfer catalysis is based on the formal deinsertion of low-valent main-group fragments via α-elimination. The first catalytic reaction based on this reactivity was reported from these laboratories, as the basis for stannane dehydropolymerizations to polystannanes.9 In this process, a d0metal hydride acts as a catalyst for dehydrogenation of the stannane, by way of a mechanism involving dehydrocoupling of the hydride with a stannane, to produce a M–SnHR2 species, and this is followed by α-H elimination of a stannylene to regenerate the hydride (Scheme 1, E = Sn). Coupled to this cycle is a polymerization involving rapid insertions of the stannylene units into M–Sn bonds.9 More recently, related dehydrocoupling reactions based on α-stibinidene (:SbR) and α-arsinidene (:AsR) elimination have been reported.10
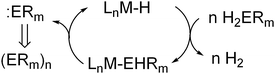 | ||
| Scheme 1 Key α-elimination step in As, Sb and Sn dehydrocoupling catalysis. | ||
Such eliminations therefore present interesting possibilities for catalytic reactions involving main-group compounds, but currently the elimination reaction appears to be limited to the heavier main group elements, for which the low-valent elimination products are expected to be most stable.
The investigation described here was designed to explore phosphinidene transfer chemistry involving a d0hafnocene fragment. In particular, phosphido alkyl complexes of the type CpCp*HfMe(PHR) were targeted as potential phosphinidene-transfer reagents. Initially, it seemed that such reactivity might arise via elimination of methane to form a phosphinidene complex; however, α-elimination of a :PR phosphinidene species (to generate CpCp*HfMe(H)) also seemed possible. The results presented here show that the dominant reactivity mode for CpCp*HfMe(PHPh) involves loss of methane and formation of a phosphinidene complex. The latter species may be employed in phosphinidene-transfer reactions, including transfers to other metal centers to produce new phosphinidene complexes of platinum and tantalum.
Results and discussion
Treatment of CpCp*HfMe(OTf) (1) with LiPHPh gives the hafnium phosphido complex CpCp*HfMe(PHPh) (2) in 73% yield as analytically pure yellow–orange crystals (eqn (1)).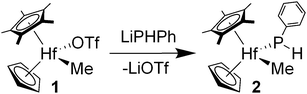 | (1) |
Characterization of 2 followed from 1H, 13C and 31P NMR and IR spectroscopy. Diagnostic features of 2 include a terminal phosphido resonance at δ −2.8 in the 31P NMR spectrum and a phosphido proton at δ 3.45 with JPH = 198 Hz and weak coupling (JHH = 1.5 Hz) to the methide ligand protons in the 1H NMR spectrum. The former value is typical of primary phosphido ligands supported by hafnocene complexes.11 Single crystals of 2 were grown from concentrated Et2O solution for an X-ray diffraction study. The molecular structure of 2, shown in Fig. 1, reveals a terminal phosphido ligand with Hf–P = 2.641(2) Å. The dihedral angle involving the Cp centroid, Hf, P and C(31) is 54°. This value shows that the phosphorus lone pair is oriented appropriately for overlap with a vacant orbital of π-symmetry at hafnium.12 The structure of 2 is most similar to that of Cp*2Hf(H)(PHPh), reported by Hillhouse and co-workers.11b Ligand-to-metal π-donation for phosphido ligands has previously been described for group 4 metallocene derivatives bearing terminal phosphido ligands.13
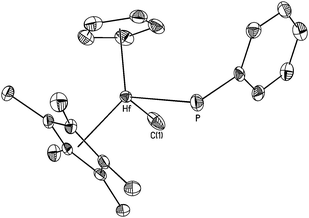 | ||
| Fig. 1 Molecular structure of CpCp*HfMe(PHPh) (2) with thermal ellipsoids shown at the 35% probability level. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Hf–C(1) 2.234(7), Hf–P 2.641(2), Hf–Cpcent 2.204(6), Hf–Cp*cent 2.194(6), P–C(31) 1.829(6); C(1)–Hf–P 97.4(2), C(31)–P–Hf 104.8(2). | ||
Heating benzene solutions of phosphide complex 2 furnishes CpCp*Hf(P3Ph3) (3) as analytically pure orange crystals in 29% isolated yield. The 31P NMR spectrum of 3 displays a doublet at δ 63.6 and a triplet at δ −158.9 with JPP = 305 Hz, indicative of a triphosphanato ligand.11a,14 The balance of the hafnium in the reaction was largely insoluble byproducts, and soluble species could not be identified. Observation of the reaction by 1H and 31P NMR spectroscopy demonstrated the liberation of methane and loss of hafnium-containing species. While the yield of 3 is low with respect to hafnium, it is noteworthy that most of the phosphorus in the reaction mixture is conserved.
The yield of triphosphanato complex 3 was improved by thermolysis of hafnium phosphide 2 with an excess (10 equiv) of PH2Ph, which afforded 3 in 92% isolated yield (eqn (2)). A small amount of P5Ph5 was observed in the reaction mixture (by 31P NMR spectroscopy), but extended heating times gave reliably low yields of the cyclic phosphine. The addition of phosphine presumably traps an intermediate, terminal phosphinidene complex similar to that suggested by Stephan in the zirconocene-catalyzed dehydrocoupling of phosphines.7Triphosphanato complexes have been known for some time, but the syntheses have often been serendipitous or characterized by low isolated yields.11a,14
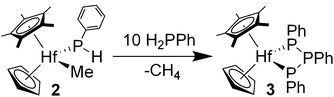 | (2) |
The thermal decomposition of 2 in benzene-d6 at 78 °C follows first-order kinetics over ca. 5 half-lives with k = 8.39(5) × 10−5s−1, as monitored by 1H NMR spectroscopy. Added PhPH2 did not affect this first-order rate, which suggests that methane loss is the key initial step in this transformation. The deuterated derivative CpCp*HfMe(PDPh) (2-dd) was analogously prepared from LiPDPh and CpCp*HfMe(OTf) and isolated as yellow crystals in 75% yield. Decomposition of 2-dd also obeys first-order kinetics with a rate k = 7.47(5) × 10−5 s−1 to give an isotope effect of kH/D = 1.12. An Eyring analysis provided activation parameters of ΔH‡ = 18.9(1) kcal mol−1 and ΔS‡ = −17.2(1) J K−1 cal−1 for T = 22.1–77.9 °C. These activation parameters are consistent with an ordered transition state. However, the value for the kinetic isotope effect is lower than might be expected for a roughly linear transfer of H from phosphorus to carbon,15 but the implications are difficult to assess in absence of more detailed knowledge of the mechanism of elimination (e.g., the potential role of a σ-complex in an equilibrium rather than kinetic isotope effect16).
Trapping reactions of CpCp*Hf![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) PPh
PPh
During the thermolysis of phosphido complex 2, methane was observed, and thermolysis of 2-dd afforded methane-d1 by 1H NMR spectroscopy. This observation suggested the initial formation of the intermediate phosphinidene complex CpCp*HfPPh. Stephan and co-workers have demonstrated that related three-coordinate zirconium phosphinidene intermediates are trapped by various organic reagents. In particular, coordination of a neutral Lewis base allows for isolation of terminal phosphinidene complexes.5b,17
Heating phosphido complex 2 with 10 equiv. of PMe3 in benzene-d6 at 75 °C for greater than 12 h resulted in formation of triphosphanato 3. Monitoring the reaction by NMR spectroscopy gives evidence for a phosphinidene intermediate, tentatively formulated as CpCp*(Me3P)HfPPh (4, Scheme 2). The most diagnostic features of this complex in the 31P NMR spectrum are resonances at δ 643.2 for the phosphinidene ligand and δ −8.6 for the PMe3 ligand, with JPP = 7 Hz. All attempts to isolate a phenylphosphinidene complex from this reaction were unsuccessful. Heating benzene solutions of phosphido complex 2 in the presence of 2-butyne resulted in methane liberation and formation of the phosphametallacyclobutene CpCp*Hf[η2-P,C:P(Ph)C(Me)C(Me)] (5), isolated as analytically pure yellow crystals in 67% yield (Scheme 2). The secondary phosphido ligand of 5 resonates at δ 53.2 in the 31P NMR spectrum, and the infrared spectrum of 5 displays a νCC stretching frequency at 1641 cm−1. The formation of 5 has precedent in observed [2 + 2]-cycloaddition reactions of zirconocene phosphinidene complexes with alkynes that spectroscopically compare well with 5.1d There is additional precedent from [2 + 2]-cycloaddition reactions of titanium phosphinidene complexes with alkynes,8 and analogous reactions of nickel complexes with alkynes and alkenes.18 The preparation of 5 is noted here as support for a transient phenylphosphinidene complex.
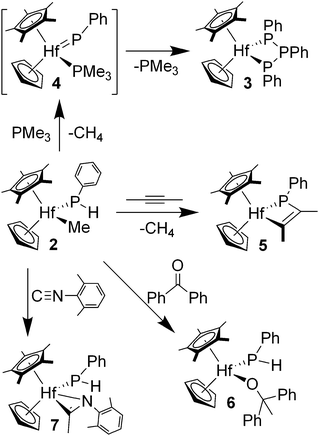 | ||
| Scheme 2 Trapping reactions of CpCp*HfMe(PHPh) (2). | ||
Other attempts to trap the hafnium phosphinidene intermediate were not successful due to competing reactions of the hafnium methyl ligand of 2. For example, reaction of 2 with benzophenone at ambient temperature in benzene gave the insertion product CpCp*Hf(PHPh)(OCMePh2) (6, Scheme 2). Analytically pure orange crystals of complex 6 were isolated as a mixture of diastereomers resulting from the presence of two chiral centers (at Hf and P). The major isomer, with a phosphido resonance at δ 4.8 in the 31P NMR spectrum, constitutes approximately 66% of the mixture, while the minor isomer possesses a phosphido resonance at δ 9.7 in the 31P NMR spectrum.
Treatment of phosphide 2 with xylyl isocyanide in benzene at ambient temperature afforded the insertion product CpCp*Hf(PHPh)[η2-C(Me)N(2,6-Me2C6H3)] (7) as analytically pure, colorless crystals in 92% isolated yield (Scheme 2). Complex 7 displays a single set of resonances in 1H, 13C and 31P NMR spectra. Noteworthy spectroscopic features of 7 include a phosphido resonance at δ −68.0 in the 31P NMR spectrum and an infrared νCN stretch of 1573 cm−1, which is consistent with an η2-iminoacyl ligand.19
These insertion reactions are notable in that related Cp′2ZrMe(PHR) complexes undergo analogous insertion reactionsvia rupture of the Zr–P (rather than the Zr–C) bond.20
Synthesis of a terminal phosphinidene complex of hafnium
A related, more stable hafnium phosphinidene derivative was sought for comparison with transient complex 4. Treatment of triflate complex 1 with LiPH(dmp)·Et2O (dmp = 2,6-dimesitylphenyl) in the presence of ca. 6 equiv. of PMe3 in toluene solution gave analytically pure dark red crystals of CpCp*(Me3P)HfP(dmp) (8) in 71% yield (Scheme 3). This synthesis is analogous to those used in the preparations of Cp2(Me3P)ZrP(Mes*) (Mes* = 2,4,6-tBu3C6H2), Cp*2(Me3P)ZrP(dmp) and Cp2(PhMe2P)ZrPPtBu,6a,21 and likely involves formation of a terminal phosphido complex which then undergoes methane elimination. Phosphinidene complex 8 was characterized by 1H, 13C and 31P NMR and IR spectroscopy. Key features of 8 include a resonance at δ 626.9 for the terminal phosphinidene ligand, and a resonance at δ −9.2 for the PMe3 ligand with JPP = 6 Hz in the 31P NMR spectrum. These data are very similar to those for transient phenylphosphinidene 4, lending additional support to that formulation.
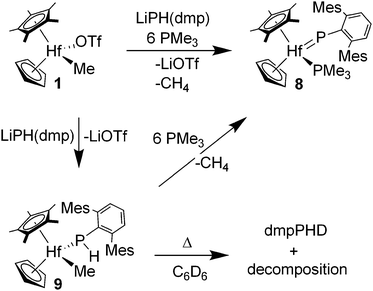 | ||
| Scheme 3 Synthesis of CpCp*(Me3P)HfP(dmp) (8). | ||
Reaction of triflato complex 1 with LiPH(dmp)·Et2O gave proposed intermediate CpCp*HfMe(PHdmp) (9) as orange crystals in 62% yield (Scheme 3). Complex 9 was characterized by NMR (1H, 13C and 31P) and IR spectroscopy. In the presence of ca. 6 equiv. of PMe3 in benzene-d6, phosphido 9 gave a 90% conversion to phosphinidene 8 with liberation of methane as observed by 1H and 31P NMR spectroscopy. Besides being thermally sensitive to methane loss, complex 9 undergoes Hf–P bond cleavage. Thus, benzene-d6 solutions of 9 decompose to (dmp)PHD and insoluble Hf-products over a period of 2 d at ambient temperature.
Treatment of CpCp*HfCl2 (10) with LiPH(dmp)·Et2O followed by 1 equiv. of methyllithium afforded analytically pure orange crystals of [CpCp*(Cl)HfP(dmp)][Li(Et2O)] (11) in 67% yield (eqn (3)). Gas evolved from the reaction mixture is presumed to be methane. The high solubility of 11 in relatively non-polar solvents such as benzene suggests a tight ion pair. Characterization of 11 followed from NMR (1H, 13C and 31P) and IR spectroscopy. A lithium test confirmed the presence of Li in the isolated material. However, a 7Li NMR spectrum of 11 could not be obtained. The mesityl substituents of the dmp group are inequivalent in the 1H and 13C NMR spectra, and the chemical shifts of one ring are consistent with a mesityl ring coordinated to lithium. Additionally, the diethyl ether in 11 appears to be coordinated to Li on the basis of 1H and 13C NMR spectra. The phosphinidene ligand of 11 resonates at δ 150.7 in the 31P NMR spectrum. This value may reflect interaction between phosphorus and the lithium ion, but no P–Li coupling was resolved in the 31P NMR spectrum. 1H and 31P NMR spectra exhibited no evidence for one-bond P–H coupling, consistent with a deprotonated phosphorus center. A related zirconocene complex, [Cp*2(Cl)ZrP(2,4,6-tBu3C6H2)][Li(dme)] has been observed by Stephan and co-workers but was not successfully isolated.5a Mindiola and co-workers have fully characterized related scandium “ate” complexes with phosphinidene ligands.3
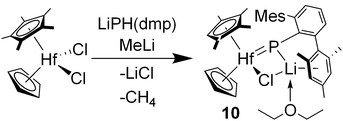 | (3) |
Phosphinidene ligand exchange
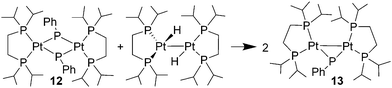
Heating benzene solutions of hafnium phosphido 2 with one equiv. of (dippe)PtCl2 (dippe = 1,2-bis(diisopropylphosphino)ethane) at 76 °C for 8 h resulted in a phosphinidene ligand transfer reaction (eqn (4)). Fractional crystallization of the product mixture gave analytically pure orange crystals of [(dippe)Pt(μ-PPh)]2 (12) and the known hafnocene dichloride 11 in 59 and 71% yields, respectively. Observation of the reaction in benzene-d6 solution by 1H and 31P NMR spectroscopy revealed quantitative conversion to 12. Platinum phosphinidene complex 12 was characterized by 1H, 13C, 31P NMR and IR spectroscopy. Central to the spectroscopic characterization of 12 are the multiplet resonances for the dippe and phosphinidene phosphorus nuclei at δ 82.8 and δ −51.6, respectively, in the 31P NMR spectrum. The phosphorus nuclei exhibit relatively small scalar couplings of JPP = 30.7 and 35.1 Hz. Another feature of note is the very small coupling between the phosphinidene phosphorus and platinum nuclei, JPtP = 827 Hz. These spectroscopic features are very similar to those of the bridging phosphinidene complex [(dppe)Pt(μ-PMes)]2 (dppe = 1,2-bis(diphenylphosphino)ethane, Mes = 2,4,6-Me3C6H2) reported by Kourkine and Glueck.22 It is expected that complex 12 does not engage in ligand to metal π-bonding and that the phosphinidene phosphorus is pyramidal. Suitable crystals for an X-ray diffraction study were not obtained.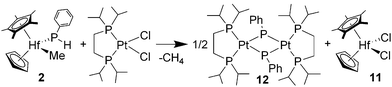 | (4) |
Transition-metal phosphinidene complexes frequently engage in chemistry analogous to phospha-Whittig reagents, such as reactions of a MPR moiety with aldehydes or ketones to give the corresponding phosphaalkene and metal oxo.1a In addition, a phosphinidene ligand can be displaced from a metal with organic dihalides to give a variety of organophosphorus products and metal dichloride salts. Both types of phosphinidene-transfer reaction have been investigated for zirconocene phosphinidene complexes by Stephan and co-workers.1d The aforementioned phosphinidene ligand exchange reaction involving two metal centers is a rare transformation.3
A crystalline derivative of μ-phosphinidene complex 12 was obtained by reaction of 12 with [(dippe)PtH]2, a known Pt(0) source,23 in benzene-d6. This gave X-ray quality yellow crystals of the phosphinidene-bridged platinum dimer [(dippe)Pt]2(μ-PPh) (13) in 54% yield (eqn (5)). Spectroscopic data for 13 is comparable to that of 12 with a phosphinidene resonance at δ −38.6 with a small JPtP value of 887 Hz. A single-crystal X-ray diffraction study confirmed the spectroscopic assignment of 13, and the molecular structure is shown in Fig. 2. An important feature of this structure is confirmation of the pyramidal phosphinidene phosphorus, as suggested by spectroscopic data (sum of angles at P(5) ∼290°). The μ-phosphinidene has bonds to platinum, Pt(1)–P(5) 2.299(3) Å, Pt(2)–P(5) 2.309(3) Å, that are similar in length to those involving the dippe ligand; Pt–P(dippe)avg values of ∼2.38 Å are consistent with a bond order of one.
 | (5) |
![Molecular structure of [(dippe)Pt]2(μ-PPh) (12) with thermal ellipsoids shown at the 35% probability level. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Pt(1)–P(5) 2.299(3), Pt(2)–P(5) 2.309(3), Pt(1)–Pt(2) 2.7396(9), P(5)–C(51) 1.84(1); Pt(1)–P(5)–Pt(2) 53.36(8), P(4)–Pt(2)–P(3) 88.2(1), P(1)–Pt(1)–P(2) 88.2(1).](/image/article/2011/SC/c1sc00119a/c1sc00119a-f2.gif) | ||
| Fig. 2 Molecular structure of [(dippe)Pt]2(μ-PPh) (12) with thermal ellipsoids shown at the 35% probability level. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Pt(1)–P(5) 2.299(3), Pt(2)–P(5) 2.309(3), Pt(1)–Pt(2) 2.7396(9), P(5)–C(51) 1.84(1); Pt(1)–P(5)–Pt(2) 53.36(8), P(4)–Pt(2)–P(3) 88.2(1), P(1)–Pt(1)–P(2) 88.2(1). | ||
Metathesis reactions of (dippe)PtCl2 with 1 or 2 equiv. of LiPHPh or with 1 equiv. of Li2PPh in diethyl ether failed to give clean reaction products, and complexes 12 or 13 were not identified in the reaction mixtures by 31P NMR spectroscopy. A synthetic route similar to that used for [(dppe)Pt(μ-PMes)]2 was not attempted.22
A terminal phosphinidene complex was targeted as a product of ligand transfer from 2. For this purpose, the new tantalum complex trans-[N(Np)Ar]3TaCl2 (14; Np = neopentyl, Ar = 3,5-dimethylaryl) was prepared as analytically pure yellow crystals by reaction of TaCl5 with 3 equiv. of LiN(Np)Ar24 in diethyl ether (Scheme 4). Spectroscopic (1H and 13C NMR; IR) features for 14 are unremarkable but support the formulation assigned to this complex. This assignment is further supported by related tantalum derivatives prepared by Cummins and co-workers.25
![Synthesis of [N(Np)Ar]3TaPPh (15).](/image/article/2011/SC/c1sc00119a/c1sc00119a-s4.gif) | ||
| Scheme 4 Synthesis of [N(Np)Ar]3TaPPh (15). | ||
Heating benzene solutions of hafnium phosphido 2 with one equiv. of 14 at 76 °C for 8 h resulted in formation of CpCp*HfCl2 (11) in 78% isolated yield and analytically pure orange crystals of terminal phosphinidene complex [N(Np)Ar]3TaPPh (15) in 84% yield (Scheme 4). The 31P NMR spectrum of complex 15 displays a resonance at δ 415.6 assigned to the terminal phosphinidene ligand. The 1H NMR spectrum of phosphinidene 15 is simple with pseudo C3-symmetry at all temperatures explored (−90–80 °C, toluene-d8). Attempts to prepare phosphinidene 15 by reaction of dichloride complex 14 with Li2PPh or two equiv. of LiPHPh gave 15 in low isolated yields from complex mixtures.
Tantalum complexes with terminal phosphinidene ligands are well known. A family of compounds of the type [N3N]TaPR (R = Ph, alkyl or silyl; [N3N]3− = [(Me3SiNCH2CH2)3N]3−) have been reported by Schrock and co-workers with phosphinidene chemical shifts in the range of δ 157–273.26 These chemical shift values are lower than that for complex 15, which may be a result of the lack of an amine ligand trans to the phosphinidene moiety in complex 15. A four-coordinate Ta-phosphinidene complex (tBu3SiO)3TaPPh, reported by Wolczanski and co-workers, has a 31P NMR resonance at δ 334.6.27 Wolf and Hey-Hawkins have reported a Cp*-supported complex, Cp*TaPh(P6Ph5), that may feature phosphinidene-like bonding.28 Finally, a highly related N(Np)Ar-supported niobium complex featuring a diphenylphosphinylphosphinidene ligand was described by Figueroa and Cummins.29
Conclusions
The reactions described above demonstrate that CpCp*HfMe(PHPh) (2) acts as a phosphinidene source, via the key intermediate CpCp*HfPPh, which is formed by a thermally-induced α-H abstraction and methane loss. Effective trapping of the phosphinidene intermediate was achieved by reaction with 2-butyne to give the [2 + 2]-cycloaddition product CpCp*Hf[η2-P,C:P(Ph)C(Me)C(Me)] (5). Further evidence for the intermediacy of CpCp*HfPPh was provided by observation of CpCp*(Me3P)HfPPh (4), and its comparison to isolated CpCp*(Me3P)HfP(dmp) (8). The most interesting reactivity associated with 2 is its ability to undergo phosphinidene ligand exchange with transition-metal dichloride complexes. Thus, reaction of 2 with (dippe)PtCl2 or [N(Np)Ar]3TaCl2 gives the new phosphinidene complexes [(dippe)Pt(μ-PPh)]2 (12) and [N(Np)Ar]3TaPPh (15). These phosphinidene complexes were readily separated from the CpCp*HfCl2 byproduct and were difficult to synthesize by alternative means.
Acknowledgements
This work was funded by the US National Science Foundation and by the Miller Institute for Basic Research in Science through a Research Fellowship to R. W.Notes and references
- (a) K. Lammertsma, Top. Curr. Chem., 2003, 229, 95–119 CAS; (b) K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102–2113 CrossRef; (c) R. Waterman, Dalton Trans., 2009, 18–26 RSC; (d) D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 315–329 CAS; (e) A. H. Cowley, Acc. Chem. Res., 1997, 30, 445–451 CrossRef CAS; (f) A. H. Cowley and A. R. Barron, Acc. Chem. Res., 1988, 21, 81–87 CrossRef CAS; (g) F. Mathey, Dalton Trans., 2007, 1861–1868 RSC; (h) S. Shah and J. D. Protasiewicz, Coord. Chem. Rev., 2000, 210, 181–201 CrossRef CAS.
- U. J. Kilgore, H. Fan, M. Pink, E. Urnezius, J. D. Protasiewicz and D. J. Mindiola, Chem. Commun., 2009, 4521–4523 RSC.
- B. F. Wicker, J. Scott, J. G. Andino, X. Gao, H. Park, M. Pink and D. J. Mindiola, J. Am. Chem. Soc., 2010, 132, 3691–3693 CrossRef CAS.
- K. Lammertsma and M. J. M. Vlaar, Eur. J. Org. Chem., 2002, 1127–1138 CrossRef CAS.
- (a) Z. Hou, T. L. Breen and D. W. Stephan, Organometallics, 1993, 12, 3158–3167 CrossRef CAS; (b) Z. Hou and D. W. Stephan, J. Am. Chem. Soc., 1992, 114, 10088–10089 CrossRef CAS.
- (a) T. L. Breen and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 11914–11921 CrossRef CAS; (b) T. L. Breen and D. W. Stephan, Organometallics, 1996, 15, 4223–4227 CrossRef CAS; (c) T. L. Breen and D. W. Stephan, J. Am. Chem. Soc., 1996, 118, 4204–4205 CrossRef CAS.
- (a) M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 12645–12646 CrossRef CAS; (b) R. Waterman, Curr. Org. Chem., 2008, 12, 1322–1339 CrossRef CAS.
- G. Zhao, F. Basuli, U. J. Kilgore, H. Fan, H. Aneetha, J. C. Huffman, G. Wu and D. J. Mindiola, J. Am. Chem. Soc., 2006, 128, 13575–13585 CrossRef CAS.
- (a) N. R. Neale and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 3802–3803 CrossRef CAS; (b) N. R. Neale and T. D. Tilley, Tetrahedron, 2004, 60, 7247–7260 CrossRef CAS; (c) N. R. Neale and T. D. Tilley, J. Am. Chem. Soc., 2005, 127, 14745–14755 CrossRef CAS; (d) J. Guihaumé, C. Raynaud, O. Eisenstein, L. Perrin, L. Maron and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 1816–1819 CrossRef CAS.
- (a) A. J. Roering, J. J. Davidson, S. N. MacMillan, J. M. Tanski and R. Waterman, Dalton Trans., 2008, 4488–4498 RSC; (b) R. Waterman and T. D. Tilley, Angew. Chem., Int. Ed., 2006, 45, 2926–2929 CrossRef CAS.
- (a) B. L. Benac and R. A. Jones, Polyhedron, 1989, 8, 1774–1777 CrossRef CAS; (b) G. A. Vaughan, G. L. Hillhouse and A. L. Rheingold, Organometallics, 1989, 8, 1760–1765 CrossRef CAS.
- J. W. Lauher and R. Hoffmann, J. Am. Chem. Soc., 1976, 98, 1729–1742 CrossRef CAS.
- R. T. Baker, J. F. Whitney and S. S. Wreford, Organometallics, 1983, 2, 1049–1051 CrossRef CAS.
- (a) E. Hey, S. G. Bott and J. L. Atwood, Chem. Ber., 1988, 121, 561–563 CrossRef CAS; (b) J. Ho, T. L. Breen, A. Ozarowski and D. W. Stephan, Inorg. Chem., 1994, 33, 865–870 CrossRef CAS.
- (a) C. M. Fendrick and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 425–437 CrossRef CAS; (b) M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203–219 CrossRef CAS; (c) T. D. Tilley, Comments Inorg. Chem., 1990, 10, 37–51 CAS; (d) H. G. Woo, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1992, 114, 5698–5707 CrossRef CAS.
- (a) W. D. Jones, Acc. Chem. Res., 2003, 36, 140–146 CrossRef CAS; (b) G. Parkin, Acc. Chem. Res., 2009, 42, 315–325 CrossRef CAS.
- E. Urnezius, S. J. Klippenstein and J. D. Protasiewicz, Inorg. Chim. Acta, 2000, 297, 181–190 CrossRef CAS.
- (a) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 CrossRef CAS; (b) R. Waterman and G. L. Hillhouse, Organometallics, 2003, 22, 5182–5184 CrossRef CAS.
- F. H. Elsner, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Organomet. Chem., 1988, 358, 169–183 CrossRef CAS.
- T. L. Breen and D. W. Stephan, Organometallics, 1996, 15, 4509–4514 CrossRef CAS.
- (a) E. Urnezius, K.-C. Lam, A. L. Rheingold and J. D. Protasiewicz, J. Organomet. Chem., 2001, 630, 193–197 CrossRef CAS; (b) J. Pikies, E. Baum, E. Matern, J. Chojnacki, R. Grubba and A. Robaszkiewicz, Chem. Commun., 2004, 2478 RSC.
- I. V. Kourkine and D. S. Glueck, Inorg. Chem., 1997, 36, 5160–5164 CrossRef CAS.
- D. J. Schwartz and R. A. Andersen, J. Am. Chem. Soc., 1995, 117, 4014–4025 CrossRef CAS.
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020–4021 CrossRef CAS.
- M. A. Rankin and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 10021–10023 CrossRef CAS.
- (a) C. C. Cummins, R. R. Schrock and W. M. Davis, Angew. Chem., 1993, 105, 758–761 CrossRef CAS; (b) J. S. Freundlich, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1996, 118, 3643–3655 CrossRef CAS.
- J. B. Bonanno, P. T. Wolczanski and E. B. Lobkovsky, J. Am. Chem. Soc., 1994, 116, 11159–11160 CrossRef CAS.
- (a) U. Segerer, R. Felsberg, S. Blaurock, G. A. Hadi and E. Hey-Hawkins, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 144, 477–480 Search PubMed; (b) R. Wolf and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2006, 1348–1351 CrossRef CAS.
- J. S. Figueroa and C. C. Cummins, Angew. Chem., Int. Ed., 2004, 43, 984–988 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental data and representative kinetic plots (PDF) and crystallographic data (CIF). CCDC reference numbers 815160 and 815161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00119a |
| ‡ Current address: Department of Chemistry, University of Vermont, Burlington, VT 05401, USA. |
| This journal is © The Royal Society of Chemistry 2011 |