Organometallic chemistry of extended periodic π-electron systems: hexahapto-chromium complexes of graphene and single-walled carbon nanotubes†
Santanu
Sarkar
,
Sandip
Niyogi
,
Elena
Bekyarova
and
Robert C.
Haddon
*
Departments of Chemistry and Chemical and Environmental Engineering, and Center for Nanoscale Science and Engineering, University of California, Riverside, California 92521, USA. E-mail: haddon@ucr.edu
First published on 4th May 2011
Abstract
We explore the organometallic chemistry of graphitic materials and demonstrate the η6-complexation reactions of chromium with graphene, graphite and carbon nanotubes. All of these extended periodic π-electron systems exhibit some degree of reactivity toward the reagents Cr(CO)6 and (η6-benzene)Cr(CO)3, and we are able to demonstrate the formation of (η6-arene)Cr(CO)3 or (η6-arene)2Cr, where arene = single-walled carbon nanotubes (SWNT), exfoliated graphene (XG), epitaxial graphene (EG) and highly-oriented pyrolytic graphite (HOPG). We report the ATR-IR, Raman spectroscopy, XPS and chemistry of these new organometallic species and we observe clearly understandable trends in the chemistry and stability of the complexes based on curvature and surface presentation. For example, the SWNTs are the least reactive presumably as a result of the effect of curvature on the formation of the hexahapto bond; in the case of HOPG, (η6-HOPG)Cr(CO)3 was isolated while the exfoliated graphene samples were found to give both (η6-graphene)2Cr, and (η6-graphene)Cr(CO)3 structures. We report simple and efficient routes for the mild decomplexation of the graphene–chromium complexes which appears to restore the original pristine graphene state; exposure of the samples to white light in a solution of acetonitrile or the use of selected ligand competition reactions bring about a clean reversal of the metal complexation reactions and provide an independent proof of structure for the reaction products. This study represents the first example of the use of graphene as a ligand and is expected to expand the scope of graphene chemistry in connection with the application of this material in organometallic catalysis, where graphene can act as an electronically conjugated catalyst support.
Introduction
Graphene, a two-dimensional crystal of atomic thickness, has attracted significant attention in recent years because of its intriguing electronic structure, which provides the basis for unique physical phenomena.1–4 The electronic properties of graphene are complemented by its mechanical and thermal properties and this has served to further raise interest in the material. In order to realize the potential applications of graphene, there is a need to develop the chemistry and to tailor its physical properties.Apart from noncovalent functionalization reactions, recent reports on the chemistry of graphene include Diels–Alder reactions,5 and covalent bond forming reactions with aryl groups,6–8 which occur under very mild conditions and may provide either a monolayer coverage,6,9 or multilayer structures of various thicknesses.10 The traditional derivatives, graphene oxide,11–20graphane (completely hydrogenated graphene)21,22 and fluorographene ((CF)n)23–27 have also been studied. In these reactions the sp2 hybridized carbon atoms of graphene are converted to sp3 hybridization; breaking the π-conjugation length in this manner is a route for band gap engineering of graphene.9,28 The flat π-surface of graphene together with its unique Fermi level electronic structure suggests the exploration of chemical reactions involving other bonding configurations. In hexahapto-metal complexation reactions, the vacant d-orbitals of a transition metal can overlap with and accept electron density from the occupied π-orbitals of graphene, resulting in the coordination of the metal atom to individual benzenoid aromatic rings of graphene and in bis-graphene metal sandwich complexes it is possible to electronically conjugate adjacent graphene sheets. The η6-metal bond to graphene is not expected to significantly rehybridize the sp2carbon atoms and can be anticipated to provide a much milder modification of the electronic structure of graphene than σ-bond formation; as a result, the metal coordination site on the graphene surface is expected to be mobile.
In prior work, η2-complexation of single-walled carbon nanotubes (SWNTs) was explored in reactions with Vaska's complex, [Ir(CO)Cl(PPh3)2]29,30 and Wilkinson's complex, [RhCl(PPh3)2]30,31 The Rh-based SWNT-Wilkinson's complex showed recyclable catalytic hydrogenation of cyclohexene at room temperature.31Chromium complexes of HiPCO single-walled carbon nanotubes have also been discussed.32 In general, the curvature of the conjugated carbon network enhances the reactivity of the fullerenes and carbon nanotubes (where addition chemistry predominates),33,34 including the dihapto-metal complexation reactions to give coordination compounds such as η2-π complexes of C60, namely η2-C60, μ-η2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :η2-C60, μ3-η2:η2:η2-C60 and η2 and η1 σ–π mixed complexes, namely μ3-η1:η2:η1-C60, μ3-η1:η1:η2-C60, together with similar complexes of higher fullerenes.35 However, it has been shown that the curvature in C60 significantly inhibits the potential of the molecule to function as a ligand in pentahapto- (η5), and hexahapto- (η6) complexation reactions because the fullerene π-orbitals are directed away from the metal as a result of the rehybridization of the ring carbon atoms.36,37 Nevertheless, the curvature can be ameliorated by functionalization,37–39 and this has allowed the preparation of organometallic fullerene derivatives.38,40 In C60 the π-orbital axis vectors are directed away from the center of the respective rings and make angles of 16.3° (POAV2) to a normal to the plane of the five-membered rings and 25.8° (POAV2) to a normal to the plane of the six-membered rings; hence, hexahapto- (η6) coordination is even more strongly disfavored than pentahapto- (η5) complexation.37 However, an examination of carbon nanotube structures, which have lower curvature than the fullerenes (carbon nanotubes are curved in 1-dimension whereas fullerenes are curved in 2-dimensions),33,34 together with the previous report on the complexation of HiPCO SWNTs encouraged us to explore the organometallic reactions of electric arc (EA)-SWNTs, which have significantly larger diameter.41,42
:η2-C60, μ3-η2:η2:η2-C60 and η2 and η1 σ–π mixed complexes, namely μ3-η1:η2:η1-C60, μ3-η1:η1:η2-C60, together with similar complexes of higher fullerenes.35 However, it has been shown that the curvature in C60 significantly inhibits the potential of the molecule to function as a ligand in pentahapto- (η5), and hexahapto- (η6) complexation reactions because the fullerene π-orbitals are directed away from the metal as a result of the rehybridization of the ring carbon atoms.36,37 Nevertheless, the curvature can be ameliorated by functionalization,37–39 and this has allowed the preparation of organometallic fullerene derivatives.38,40 In C60 the π-orbital axis vectors are directed away from the center of the respective rings and make angles of 16.3° (POAV2) to a normal to the plane of the five-membered rings and 25.8° (POAV2) to a normal to the plane of the six-membered rings; hence, hexahapto- (η6) coordination is even more strongly disfavored than pentahapto- (η5) complexation.37 However, an examination of carbon nanotube structures, which have lower curvature than the fullerenes (carbon nanotubes are curved in 1-dimension whereas fullerenes are curved in 2-dimensions),33,34 together with the previous report on the complexation of HiPCO SWNTs encouraged us to explore the organometallic reactions of electric arc (EA)-SWNTs, which have significantly larger diameter.41,42
We begin by discussing the results of the reaction of EA-SWNTs with (η6–benzene)Cr(CO)3 and then move to an investigation of the reaction of various forms of graphite and graphene with Cr(CO)6 and (η6-benzene)Cr(CO)3. These two reagents were chosen to explore the fundamental arene–metal complexation mechanisms,43 which depend on the displacement of three carbon monoxide molecules and in the present manuscript we demonstrate that all of these forms of conjugated carbon exhibit some degree of reactivity, as delineated in Scheme 1. The study demonstrates the wide applicability of this type of chemistry in engineering the electronic structure of extended, periodic π-electron systems as well as expanding the scope of graphene chemistry towards applications in organometallic catalysis and electronics. Additionally, we demonstrate a number of routes for de-complexation that appear to restore graphene to its pristine state.
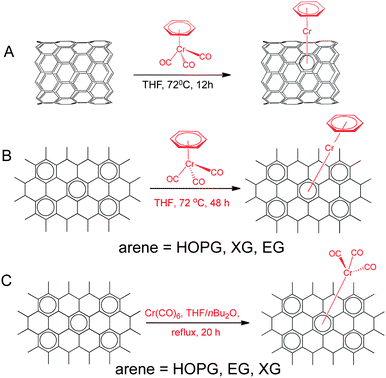 | ||
| Scheme 1 Representative reactions of SWNT, graphite and graphene with (η6-benzene)Cr(CO)3 and Cr(CO)6 based on product structures. | ||
Experimental section
Chemicals and instrumentation
Cr(CO)6 (98%, Aldrich, FW = 220.06, m.p. > 150 °C), (η6-benzene)Cr(CO)3 (98%, FW = 214.4, m.p. = 163–166 °C), ortho-dichlorobenzene (anhydrous, 99%), p-xylene (anhydrous, 99%), mesitylene (98%), microcrystalline graphite (1–2 micron, synthetic), dibutyl ether (anhydrous, 99.3%) were purchased from Sigma-Aldrich. HOPG was obtained from Union Carbide Corporation. The single-walled nanotube (SWNT) samples were obtained from Carbon Solutions Inc. (P2-SWNT, http://www.carbonsolution.com). The epitaxial graphene films grown on the C-face of SiC substrates (Cree Inc, 4H SiC) with dimensions of 3.5 × 4.5 mm2 were provided by Professor Walt de Heer.44,45The ATR-IR spectra were taken using a Thermo Nicolet Nexus 670 FTIR instrument, equipped with an ATR sampling accessory. Raman spectra were acquired in a Nicolet Almega XR Dispersive Raman microscope with a 0.6 μm spot size and 532 nm excitation. STM images were acquired under ambient conditions using Pt/Ir tips in a DI Nanoscope IIIa multimode SPM. TGA was performed at a heating rate of 5 °C min−1 in air using a Pyris 1 thermo-gravimetric analyzer (Perkin Elmer). NMR spectra were collected in a Bruker 300 MHz instrument and UV-vis spectra were collected in a Varian Cary 5000 spectrometer.
Resistivity measurements
The resistance was measured at room temperature in air with a Keithley 236 source-measure unit. The temperature dependence of resistivity was measured in a custom-made helium variable-temperature probe using a Lake Shore 340 temperature controller driven with custom LabVIEW software. The device was degassed for 12 h in vacuum (2 × 10−8 torr) prior to the measurement.XPS measurement
The XPS measurements were performed with a Kratos Axis Ultra spectrometer (Kratos Analytical, Manchester, UK) using Al Kα monochromated radiation at 1486 eV at base pressure of about 3 x10−8 Torr. The survey spectra were recorded using 270 watts of X-ray power, 80 pass energy, 0.5 eV step size. A low-energy electron flood from a filament was used for charge neutralization. The spectra are shown without energy-scale correction.Preparation of the chromium–SWNT complex, (η6-benzene)Cr(η6-SWNT)
(η6-benzene)Cr(CO)3 (17 mg, 0.08 mmol, FW = 214.4) was added to a suspension of purified SWNTs (18 mg, 1.5 mmol of carbon atoms; purified SWNTs—P2-SWNT) in dry distilled THF (6 mL). The reaction mixture was sonicated for 2 min using an ultrasonic probe (Cole-Parmer, 50% amplitude) and then degassed with argon for 15 min in absence of light. The reaction mixture was heated at 72 °C for 72 h in the dark under a positive pressure of argon, after which it was cooled to room temperature. The suspension was filtered through a 0.2 μm PTFE membrane and the solid was washed with anhydrous ether. The resulting chromium–SWNTs complex (∼21 mg, isolated yield) was dried overnight under vacuum in the dark.Exfoliation of microcrystalline graphite
Microcrystalline graphite (1–2 μm, 500 mg, synthetic, Sigma-Aldrich) was sonicated in o-dichlorobenzene (∼200 mL ODCB) for 1 h using a probe ultrasonic processor (Cole-Parmer) at 40% amplitude. The dispersion was centrifuged at 14000 g for 30 min. The resulting supernatant (which yielded dispersions of graphene in o-dichlorobenzene)46 was collected and concentrated under vacuum. The powdered exfoliated graphene was dried in high vacuum overnight and used for subsequent reactions after re-dispersion in dry, distilled THF.Reaction of exfoliated graphene and (η6-benzene)Cr(CO)3
In a typical reaction, (η6-benzene)Cr(CO)3 (36 mg, 0.17 mmol, FW = 214.4) was added to a suspension of exfoliated graphene (20 mg, 1.67 mmol of carbon) in THF (4 mL). The reaction mixture was stirred vigorously and refluxed at 72 °C under argon, in the absence of light for 48 h. The resulting mixture was filtered using 0.2 μm PTFE filter paper and the solid was washed with fresh THF and ether (to remove excess chromium reagent). The resulting solid was dried under vacuum overnight in dark to obtain a silver-colored solid (∼27 mg of solid was isolated).Reaction of exfoliated graphene and Cr(CO)6
Cr(CO)6 [8.2 (47.8) mg, 0.04 (0.22) mmol, FW = 220.06] was added to a suspension of exfoliated graphene (20 mg) in THF [4 (10) mL] and dibutyl ether [2 (5) mL]. The black suspension was stirred vigorously and refluxed at 72 °C in the absence of light under an atmosphere of argon for 48 h. The reaction mixture was filtered using 0.2 μm PTFE filter paper and the solid was washed with fresh THF and ether. The resulting solid (∼19 mg) was dried under vacuum overnight in the dark.Reaction of HOPG and EG with Cr(CO)6
HOPG (∼0.28 cm2) or EG on 4H-SiC (3.5 mm × 4.5 mm), was heated in a solution of Cr(CO)6 (30 mg, 0.14 mmol) in THF (3 mL) and dibutyl ether (1 mL) under a positive pressure of argon at 72 °C for 48 h without stirring, then washed with anhydrous ether and dried under a gentle flow of argon.Reaction of HOPG and EG with (η6-benzene)Cr(CO)3
A piece of HOPG (∼0.32 cm2) or EG on 4H-SiC (3.5 mm × 4.5 mm) was heated in a solution of (η6-benzene)Cr(CO)3 (33 mg, 0.16 mmol) in THF (3 mL) under a positive pressure of argon at 72 °C for 72 h without stirring, after which the sample was washed with THF and anhydrous ether and dried under a gentle flow of argon.De-complexation of graphene–Cr complexes by ambient oxidation
To collect the Raman spectra of the graphene–Cr complexes, a dispersion of the sample was allowed to dry on a SiO2 substrate; the color contrast with the substrate allowed identification of graphene samples of various thickness. After recording the Raman spectra of the products, the decomplexation reaction was carried out by adding a few drops of acetonitrile to the substrates and exposing them to sunlight, under a glass petridish; Raman spectroscopy was used to follow the progress of the de-complexation reaction.De-complexation with electron rich arenes
The Cr complexes of XG and HOPG were either refluxed or warmed (oil bath temperature of 100 °C for benzene, 150 °C for p-xylene and 150 °C for mesitylene) with the arene (∼5 mL) under argon overnight. The resulting reaction mixture was filtered through a 0.2 μm PTFE membrane and the solid was washed with a copious amount of anhydrous diethylether. The resulting solid was dried for 1 h under vacuum and characterized by Raman spectroscopy.Results and discussion
In metal carbonyl complexes and their ligand exchanged derivatives, the frequency of the C–O stretching vibration can be used to distinguish between terminal (νCO = 2000 ± 100 cm−1) and bridged (νCO = 1800 ± 75 cm−1) CO structures as well as providing information on the electronic environment of the metal center.47,48 In SWNT, HOPG and graphene chemistry, the use of IR spectroscopy is sometimes limited by weak spectral features as a result of the dynamic dipole moments and IR assignments of even well documented complexes can be difficult.49 In the case of metal complexes of polyaromatic arenes, haptotropic slippage50 and fluxional behavior51 of the ligands further complicates characterization using FT-IR spectroscopy.43 Scanning tunneling microscopy provides more direct evidence of chemical functionalization, if well ordered samples from optimized reactions are available.52–54 We have made use of FT-IR spectroscopy in characterizing the products of the reaction of SWNTs, HOPG, and graphene with organometallic precursors, together with Raman microscopy (excitation at λ = 532 nm) and absorbance spectroscopy which are well known in carbon materials science. In SWNTs, the inter-band transitions in metallic and semiconducting structures give rise to distinct peaks in the UV-vis-NIR absorbance spectrum and modification of the π-conjugation affects these band transitions. For example, in the side-wall functionalization of SWNTs with dichlorocarbene, the change in the absorbance spectrum was related to the degree of functionalization, showing that after complete reaction, all band transitions are lost.55 Similar effects have been demonstrated by other groups using different chemical reactions.56 In graphitic materials, the frequency, width and spectral shape of the most intense Raman peaks, labeled as the G and 2D bands, can be used to determine the number of layers, their inter-layer electronic interaction as well as the Fermi-level electronic structure due to the resonant enhancement of these vibrations.57 In graphene samples prepared using various techniques, the intensity of the D-band has been found to be a useful signature for following the effect of covalent chemical reactions.7–9,21,58 The D-band is due to the in-plane breathing vibration of conjugated six-membered rings and this feature is not observed without some disruption of the long range periodicity of the graphene lattice; in covalently functionalized graphene, an empirical relationship between the density of sp3 centers and the ratio of the intensities of the G and D bands, has been reported.9,59,60 The detailed understanding of the D-band in terms of molecular vibrations in various bonding environments is also of interest in small molecule chemistry,61–63 and the present study clearly relates to this work.Reaction of (η6-benzene)Cr(CO)3 with EA-SWNTs
The reaction of EA-SWNTs (average diameter Dav = 1.55 ± 0.1 nm),41,42 with (η6-benzene)Cr(CO)3 in tetrahydrofuran (THF), which is illustrated in Scheme 1A, gave rise to a black powder that was isolated by filtration.The changes in the Raman spectrum of SWNTs due to reaction is shown in Fig. 1A; the intensity of the D-band increases relative to the G-band as previously observed for a sidewall functionalization process;55 (ID/IG ∼ 0.04 as compared to ID/IG ∼ 0.01 in the pristine SWNTs). The SWNT radial breathing mode (RBM) is resonantly enhanced by interband electronic transitions and the frequency is inversely proportional to the diameter. Thus, when the SWNTs are chemically functionalized the band transition energies are modified and this may affect the resonance conditions in the Raman experiment and in cases where the chemical reaction is dependent on nanotube diameter and chirality the RBM band profile takes on a different shape due to changes in the resonance conditions of the various SWNT populations.64 The inset in Fig. 1A shows such a change in the RBM profile; although nanotube chiralities cannot be assigned from a single excitation Raman spectrum, the loss of intensity at the lower frequency of the RBM band indicates that the larger diameter SWNTs are preferentially removed from resonance by the chemical reaction.
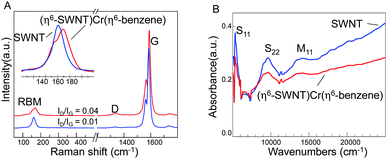 | ||
| Fig. 1 Characteristics of the (η6-SWNT)Cr(η6-benzene) complex of single-walled carbon nanotubes. A) Raman spectra of the starting SWNTs and the products, collected with λEX = 532 nm on solid samples. The inset shows the RBM region of the spectra; B) absorbance spectra of the starting SWNTs and the products, collected on dispersions in dimethylformamide; the dispersions were prepared at approximately similar optical densities and the spectra are not normalized. The features on the lower energy side of the S11 band and on both sides of the S22 bands are due to water in the solvent. | ||
The UV-vis-NIR absorption spectrum of the reaction product (Fig. 1B) shows a decrease in the intensities of all interband transitions; this is most clearly seen for the second semiconducting interband transition (S22). It is apparent that the intensities of the larger diameter SWNTs are preferentially weakened in the product in accord with the previous discussion, which suggests that lower curvature structures will be the most reactive. The change of the SWNT spectra on reaction with benzene chromium tricarbonyl is qualitatively similar to that observed on side-wall functionalization with dichlorocarbene,55 although the reaction does not proceed to the same degree, perhaps due to the incomplete dispersal of the current sample. The ATR-IR spectrum of the product (Supporting Information, Fig. S1†) does not show the C–O vibrations, but it will be of some interest to determine the mode of complexation as it is possible that chromium could bind to the interior wall of the carbon nanotubes.37
In order to examine the effect of the complexation of SWNTs with Cr on the electronic structure of the material, free-standing films of pristine SWNTs and the (η6-SWNT)Cr(η6-benzene) product were prepared and transferred to a glass substrate with pre-deposited gold contacts.65 The SWNT film thickness was estimated from the near-IR spectra of the films (absorbance at 550 nm)66–68 The conductivity of the functionalized SWNTs (σRT ∼ 100 S cm−1) decreased by a factor of 3 from the pristine value of σRT ∼ 300 S cm−1.68
Reaction of Cr(CO)6 and (η6-benzene)Cr(CO)3 with exfoliated graphene (XG)
The exfoliated graphene (XG) sample consists of a mixture of multilayer-graphene flakes (micrometer dimensions), together with single layer graphene, as evidenced by comparing the intensities of the 2D and G bands in the spectra shown in Fig. 2A and 2B and the lineshape of the 2D band.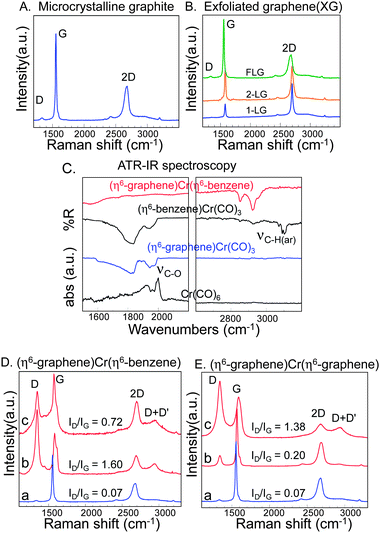 | ||
| Fig. 2 Characteristics of the exfoliated graphene samples prepared by sonication and centrifugation of microcrystalline graphite in o-dichlorobenzene and complexes. A) Raman spectrum of the starting microcrystalline graphite. B) Raman spectra of the sample after sonication in ODCB and centrifugation, showing characteristics of few layer graphene (FLG), bilayer graphene (2-LG) and single layer graphene(1-LG). C) FT-IR spectra of the starting chromium reagents and the (η6--graphene)Cr(η6-benzene) product. D) Raman spectra of the (η6-graphene)Cr(η6-benzene) product, collected on solid samples; spectra collected from different graphene flakes (spectra b and c) are shown together with that of the starting material (spectrum a); E) Raman spectra of (η6-graphene)Cr(η6-graphene) product collected on solid samples; spectra collected from different regions (spectra b and c) of the sample are shown together with that of the starting material (spectrum a). | ||
After reaction of XG with 0.13 equivalents of Cr(CO)6 (Scheme 1C) the IR spectrum showed C–O stretching vibrations at 1939 cm−1 (Fig. 2C and Supporting Information Fig. S2†), compared to those in Cr(CO)6 at 2000 cm−1. The Cr2p XPS spectra show the presence of Cr(0) (Supporting Information Fig. S3†) and the product is assigned as (η6-graphene)Cr(CO)3. After the reaction of XG with 0.02 equivalents of Cr(CO)6, the C–O vibrations could not be observed in the product (Supporting Information, Fig. S2†) but the Cr2p XPS spectra shows the presence of Cr(0) (Supporting Information Fig. S3†) and we assign the structure of the product as (η6-graphene)Cr(η6-graphene). These observations of varying product structures as a function of the reagent stoichiometry is consistent with those reported for molecular chromium complexes.69,70 The ATR-IR spectrum (Supporting Information Fig. S4†) of the reaction product of XG and (η6-benzene)Cr(CO)3 showed the aromatic C–H vibration of benzene at lower frequency compared to that in the starting material71 and does not show the C–O vibrations, indicating the formation of the (η6-graphene)Cr(η6-benzene) product (Fig. 2C).
In Fig. 2D and 2E, the changes in the Raman spectra of XG after reaction with (η6-benzene)Cr(CO)3 and Cr(CO)6 are compared. Both reactions show a strong D-band accompanied by (D + D′) peaks. The appearance of strong D peaks in the Raman spectra of the products is attributed to the chromium complexation of graphenevia η6-bond formation and associated changes in the electronic structure of graphene. The spectra signify an entirely distinct electronic structure from that of nitrophenyl-functionalized graphene, which gives sharp Raman peaks even at high coverage densities.9
Thermogravimetric analysis of (η6-graphene)Cr(η6-benzene) in air left a residue of 1.8 wt% (Supporting Information, Fig. S5†), which we attribute to the presence of chromium metal that is converted to Cr2O3 (recovered as a dark green residue) during pyrolysis and corresponds to approximately 0.2 atomic % Cr or 1 Cr atom per 500 C atoms. The low yield (metal loading) of the reaction may be due to the presence of incompletely exfoliated graphite nanoparticles;72,73 in the case of multilayer graphene particles only the outermost surfaces can participate in the complexation reaction. Thus, XPS analysis showed presence of 1.3 atomic % Cr in this material.
Regeneration of exfoliated graphene from (η6-graphene)Cr(CO)3 and (η6-graphene)Cr(η6-benzene)
The organometallic chemistry of (arene)metal(CO)3 complexes provides a number of routes for the one-step derivatization of arene molecules, which is of particular interest in the development of graphene chemistry.43,74 The simplest of these are the arene decomplexation or arene exchange reactions (Scheme 2); the arene–metal bond in (arene)Cr(CO)3 complexes is cleaved upon oxidation of the metal by exposure of the complex to sunlight in air in the presence of diethyl ether or acetonitrile, to yield the free arene.75 Refluxing (arene)Cr(CO)3 complexes in the presence of electron-rich arenes such as mesitylene and pyridine, can also yield arene exchanged Cr(CO)3 complexes.75 These decomplexation reactions are of interest because they provide a route to reverse the changes in the graphene electronic properties.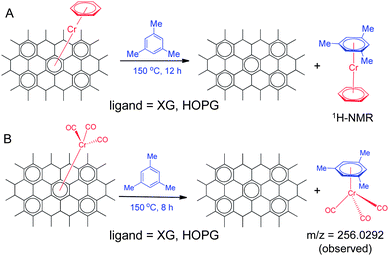 | ||
| Scheme 2 Decomplexation of graphene–chromium complexes by substitution reactions with mesitylene. | ||
Fig. 3A shows that the ratio of the intensities of the D and G bands (ID/IG) in the spectra of the (η6-graphene)Cr(η6-graphene) complex decreases to 0.06 after exposure to light for 3 h in acetonitrile (compared to ID/IG = 1.59 for the same complex before exposure to sunlight). The de-complexation of (η6-graphene)Cr(η6-benzene) requires longer exposure to sunlight; after exposure for 6 h the ID/IG ratio decreases from 1.40 to 0.18 (Fig. 3B) suggesting that the (η6-graphene)Cr(η6-graphene) complex is more reactive than (η6-graphene)Cr(η6-benzene). The strength of the (η6-graphene)–Cr bond was investigated in a series of competition reactions with electron rich arenes.76 Refluxing the (η6-graphene)Cr(η6-benzene) complex (ID/IG ∼ 1.1, Fig. 3C) in benzene (oil bath temperature 100 °C; final ID/IG = 0.85) or p-xylene (oil bath temperature 150 °C; final ID/IG = 0.45) was insufficient to fully regenerate the XG, whereas heating in mesitylene (oil bath temperature 150 °C, Scheme 2A) gave a final ID/IG = 0.04, indistinguishable from the starting graphene sample. Thus, it is apparent that the (η6-graphene)–Cr bond is fairly robust; the (η6-benzene)–Cr bond energy is reported to be 164 kJ mol−1.76
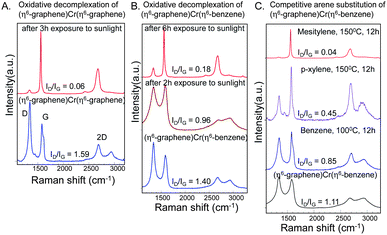 | ||
| Fig. 3 Raman characteristics of the de-complexation reactions of exfoliated graphene (EG); changes of the (η6-graphene)Cr(η6-graphene) product. (A) Effect of sunlight on (η6-graphene)Cr(η6-graphene). (B) Effect of sunlight on the (η6-graphene)Cr(η6-benzene). (C) De-complexation of (η6-graphene)Cr(η6-benzene) with benzene, p-xylene and mesitylene. | ||
The product from the decomplexation reaction of (η6-graphene)Cr(η6-benzene) in mesitylene was yellow in color, turned green on standing in air and eventually yielded a dark green precipitate (Supporting Information, Fig. S6†). The yellow extract in mesitylene was characterized using 1H NMR spectroscopy (Supporting Information Fig. S7†); based on the changes in the chemical shifts of the aromatic protons and their integration values, the decomplexed product is assigned to (η6-mesitylene)Cr(η6-benzene). The product was also analyzed using UV-vis absorption spectroscopy (Supporting Information Fig. S8†) which showed charge transfer bands between arene and chromium in the yellow solution and FAB-MS showing a m/z = 282.2862 (Supporting Information, Fig. S9†).
Reaction of Cr(CO)6 and (η6-benzene)Cr(CO)3 with epitaxial graphene (EG) and highly oriented pyrolytic graphite (HOPG)
Compared to the high surface area exfoliated graphene (XG), HOPG and epitaxial graphene are millimeter scale samples with long range crystalline order and well defined surfaces; their distinguishing feature is the interlayer stacking pattern. In HOPG, the graphene layers are Bernal stacked with strong interlayer electronic interaction, resulting in the formation of two triangular sublattices in the AB plane as seen in the STM image in Fig. 4A. In epitaxial graphene grown on SiC wafers, the graphene layers are rotationally disordered, eliminating the periodic electronic interaction in the c-axis direction and the STM image of the surface shows the typical (√3 × √3)R30° reorganization (Fig. 4B). Beyond the development of chemistry to modify the physical properties of graphene, it is interesting to compare the chemical consequences of interlayer stacking in epitaxial graphene and graphite.8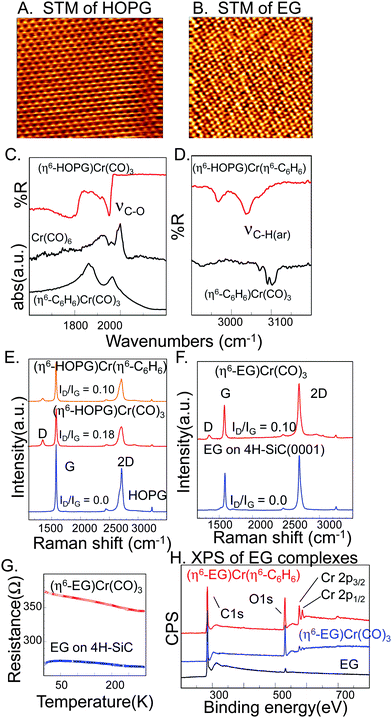 | ||
| Fig. 4 Characteristics of the complexation reactions on HOPG and epitaxial graphene. A) 5 × 3.9 nm STM image of HOPG collected under ambient conditions, Vs = +5 mV, It = 2 nA. B) 5 × 5 nm STM image of epitaxial graphene, Vs = +8 mV, It = 1 nA. C) and D) ATR-IR spectra of the chromium reagents and the products with HOPG. E) Raman characteristics of the reaction of Cr(CO)6 and (η6-benzene)Cr(CO)3 with HOPG. F) Raman spectra of epitaxial graphene before and after reaction with Cr(CO)6. G) Temperature dependence of sheet resistance of pristine and functionalized epitaxial graphene wafer. H) XPS of pristine EG on 4H-SiC(0001) and the complexation products with the chromium reagents. | ||
The IR C–O stretch in (η6-HOPG)Cr(CO)3 was observed at 1948 cm−1 while in the starting Cr(CO)6 complex it was observed at 2000 cm−1 and in (η6-benzene)Cr(CO)3 the C–O stretching was observed at 1942 cm−1 (Fig. 4C and Fig. S10 in Supporting Information†). It is known that, when arene ligands substitute for CO in Cr complexes, the electron density on Cr increases and the CO vibration frequency decreases.47 After reaction with (η6-benzene)Cr(CO)3 the C–O vibrations could not be identified and the aromatic C–H vibrations are observed at lower frequency than that of the reactant (Fig. 4D and Supporting Information, Fig. S10†).71
In contrast to the chromium complexes of exfoliated graphene, the Raman spectrum of the (η6-HOPG)Cr(CO)3 and (η6-HOPG)Cr(η6-benzene) showed a weak D-band with ID/IG = 0.18 and 0.10 respectively (Fig. 4E); treatment of EG with Cr(CO)6 also gave rise to very weak D-bands with ID/IG = 0.10 (Fig. 4F). The reaction of Cr(CO)6 with a pre-wired EG sample led to change in the transport properties of epitaxial graphene; the four point resistance of the EG sample increased by about 30% after treatment with Cr(CO)6 (Fig. 5G), as compared to the factor of two increase in resistance that was observed on nitrophenyl functionalization.6 The EG samples on SiC wafers do not provide a compatible configuration for ATR-IR measurements, so X-ray photoelectron spectroscopy (XPS) was used to characterize the chemical composition of the products. As shown in Fig. 4H, the survey scan of pristine EG reveals a strong C-peak at 284.5 eV corresponding to sp2carbon and a weak O1s peak presumably due to oxygen atoms bound to carbon at the graphene edges. The spectra of (η6-EG)Cr(η6-benzene) and (η6-EG)Cr(CO)3 show distinct peaks at ∼576.7 eV and ∼586.3 eV, which are assigned to Cr 2p3/2 and Cr 2p1/2, respectively.
De-complexation of the (η6-HOPG)Cr(CO)3 complex: a direct proof of structure
When the (η6-HOPG)Cr(CO)3 complex was subjected to an arene exchange reaction (Scheme 2B) in the presence of excess mesitylene, a deep yellow solution was obtained and the Raman spectrum of the HOPG substrate reverted to that of pristine HOPG. The FAB–MS of the yellow colored solution showed the presence of a compound of composition C12H12O3Cr [m/z: (M + H)+ = 257.0266] (Supporting Information, Fig. S11†), indicating that the reaction had led to the dissociation of the HOPG chromium complex with the formation of (η6-mesitylene)Cr(CO)3. The yellow solution in mesitylene turned green on standing, indicating the slow decomposition of the (η6-mesitylene)Cr(CO)3 due to aerial oxidation (similar to that observed for the (η6-graphene)Cr(η6-benzene) complex). This result, together with the shift of the C–O stretching frequency to 1948 cm−1 in the FT-IR spectra of the reaction product (compared to the appearance of C–O stretching frequency at 2000 cm−1 in Cr(CO)6) (Supporting Information, Fig. S10†) confirms the assignment of the reaction product from HOPG and Cr(CO)6, as (η6-HOPG)Cr(CO)3.Conclusions
The organometallic chemistry of graphitic compounds holds great potential for the realization of new materials and we have been able to demonstrate the η6-complexation reactions of chromium with various forms of graphene, graphite and carbon nanotubes. These extended periodic π-electron systems exhibit varying degrees of reactivity toward the reagents Cr(CO)6 and (η6-benzene)Cr(CO)3, and we report the formation of (η6-arene)Cr(CO)3 or (η6-arene)2Cr complexes, where arene = single-walled carbon nanotubes (SWNT), exfoliated graphene (XG), epitaxial graphene (EG) and highly-oriented pyrolytic graphite (HOPG). The compounds were characterized by a combination of IR, Raman, XPS, TGA and further chemical derivatization and we observe clearly understandable trends in the chemistry and stability of the complexes based on curvature and surface presentation. The SWNTs are the least reactive and the least stable, as a result of the effect of curvature on the formation of the hexahapto bond; in the case of substrates with well-defined surfaces such as HOPG and EG, we observe the formation of compounds such as (η6-HOPG)Cr(CO)3. The exfoliated graphene (XG) samples were found to give (η6-graphene)2Cr structures as a result of the accessibility of multiple graphene surfaces to chemical reaction as wells as (η6-graphene)Cr(CO)3 structures depending on the stoichiometry of the chromium reagent used. We report convenient routes for the decomplexation of the graphene–chromium complexes, which restore the original pristine graphitic surface; exposure of the samples to light or the use of selected ligand competition reactions lead to a reversal of the metal complexation reactions and provide an independent proof of structure for the reaction products. Haptotropic slippage50 and fluxional behavior51 of the polyaromatic ligand in (η6-arene)–Cr complexes is known to be dependent on the stoichiometry and relative binding affinity of the ligands,43 which suggests that the chromium (Cr) center will be mobile on graphitic surfaces. This study reports the first use of graphene as a ligand and is expected to expand the scope of graphene chemistry in connection with the applications of this material in electronics and organometallic catalysis, where graphene can act as an electronically conjugated catalyst support.Acknowledgements
We acknowledge financial support from DOD/DMEA under contract H94003-10-2-1003 and NSF-MRSEC through contract DMR-0820382. The authors thank Walt de Heer and his group for providing epitaxial graphene samples for this study. The SEM imaging was done at CFAMM-UCR and the MS analysis was done at ACIF-UCR. The XPS measurements were done at UCSB-MRL.References
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS.
- F. Miao, S. Wijeratne, Y. Zhang, U. C. Coskun, W. Bao and C. N. Lau, Science, 2007, 317, 1530 CrossRef CAS.
- S. Sarkar, E. Bekyarova, S. Niyogi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 3324 CrossRef CAS.
- E. Bekyarova, M. E. Itkis, P. Ramesh, C. Berger, M. Sprinkle, W. A. de Heer and R. C. Haddon, J. Am. Chem. Soc., 2009, 131, 1336 CrossRef CAS.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398 CrossRef CAS.
- F. M. Koehler, A. Jacobsen, K. Ensslin, C. Stampfer and W. J. Stark, Small, 2010, 6, 1125 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hick, M. Sprinkle, C. Berger, C. N. Lau, W. A. de Heer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061–4066 CrossRef CAS.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429 RSC.
- N. A. Kotov, I. Dekany and J. H. Fendler, Adv. Mater., 1996, 8, 637 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- H. P. Boehm, Carbon, 2002, 40, 145 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323 CrossRef CAS.
- X. S. Wu, M. Sprinkle, X. B. Li, F. Ming, C. Berger and W. A. de Heer, Phys. Rev. Lett., 2008, 101, 026801 CrossRef.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939 CrossRef CAS.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845 CrossRef CAS.
- P. K. Ang, S. Wang, Q. L. Bao, J. T. L. Thong and K. P. Loh, ACS Nano, 2009, 3, 3587 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS.
- S. Ryu, M. Y. Han, J. Maultzsch, T. F. Heinz, P. Kim, M. Steigerwald and L. E. Brus, Nano Lett., 2008, 8, 4597 CrossRef CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS.
- S. B. Bon, L. Valentini, R. Verdejo, J. L. G. Fierro, L. Peponi, M. A. Lopez-Manchado and J. M. Kenny, Chem. Mater., 2009, 21, 3433 CrossRef CAS.
- F. Withers, M. Dubois and A. K. Savchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 073403 CrossRef.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan and E. S. Snow, Nano Lett., 2010, 10, 3001 CrossRef CAS.
- J. Zhou, Q. F. Liang and J. M. Dong, Carbon, 2010, 48, 1405 CrossRef CAS.
- S. H. Cheng, K. Zou, F. Okino, H. R. Gutierrez, A. Gupta, N. Shen, P. C. Eklund, J. O. Sofo and J. Zhu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205435 CrossRef.
- E. Bekyarova, M. E. Itkis, P. Ramesh and R. C. Haddon, Phys. Status Solidi RRL, 2009, 3, 184 Search PubMed.
- S. Banerjee and S. S. Wong, Nano Lett., 2002, 2, 49 CrossRef CAS.
- S. Banerjee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17 CrossRef CAS.
- S. Banerjee and S. S. Wong, J. Am. Chem. Soc., 2002, 124, 8940 CrossRef CAS.
- C. Menard-Moyon, F. Dumas, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2006, 128, 14764 CrossRef CAS.
- R. C. Haddon, Science, 1993, 261, 1545 CAS.
- S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Acc. Chem. Res., 2002, 35, 1105 CrossRef CAS.
- K. Lee, H. Song and J. T. Park, Acc. Chem. Res., 2003, 36, 78 CrossRef CAS.
- J. R. Rogers and D. S. Marynick, Chem. Phys. Lett., 1993, 205, 197 CrossRef CAS.
- R. C. Haddon, J. Comput. Chem., 1998, 19, 139 CrossRef CAS.
- M. Sawamura, H. Iikura and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 12850 CrossRef CAS.
- Y. Matsuo, K. Tahara and E. Nakamura, J. Am. Chem. Soc., 2006, 128, 7154 CrossRef CAS.
- M. Sawamura, Y. Kuninobu, M. Toganoh, Y. Matsuo, M. Yamanaka and E. Nakamura, J. Am. Chem. Soc., 2002, 124, 9354 CrossRef CAS.
- M. A. Hamon, M. E. Itkis, S. Niyogi, T. Alvaraez, C. Kuper, M. Menon and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 11292 CrossRef CAS.
- F. Wang, M. E. Itkis and R. C. Haddon, Nano Lett., 2010, 10, 937 CrossRef CAS.
- G. Pampaloni, Coord. Chem. Rev., 2010, 254, 402 CrossRef CAS.
- W. A. de Heer, C. Berger, X. S. Wu, P. N. First, E. H. Conrad, X. B. Li, T. B. Li, M. Sprinkle, J. Hass, M. L. Sadowski, M. Potemski and G. Martinez, Solid State Commun., 2007, 143, 92 CrossRef CAS.
- E. Rolling, G. H. Gweon, S. Y. Zhou, B. S. Mun, J. L. McChesney, B. S. Hussain, A. Fedorov, P. N. First, W. A. de Heer and A. Lanzara, J. Phys. Chem. Solids, 2006, 67, 2172 CrossRef CAS.
- C. E. Hamilton, J. R. Lomeda, Z. Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460 CrossRef CAS.
- R. Cataliotti, A. Poletti and A. Santucci, J. Mol. Struct., 1970, 5, 215 CrossRef CAS.
- J. E. Huheey, Inorganic Chemistry: Principles of Structure and Reactivity, 1983, Third Edition, New York Search PubMed.
- U. J. Kim, X. M. Liu, C. A. Furtado, G. Chen, R. Saito, J. Jiang, M. S. Dresselhaus and P. C. Eklund, Phys. Rev. Lett., 2005, 95, 157402 CrossRef CAS.
- E. P. Kundig, C. Perret, S. Spichiger and G. Bernardinelli, J. Organomet. Chem., 1985, 286, 183 CrossRef.
- H. Amouri and M. Gruselle, Chem. Rev., 1996, 96, 1077 CrossRef CAS.
- D. Bonifazi, C. Nacci, R. Marega, S. Campidelli, G. Ceballos, S. Modesti, M. Meneghetti and M. Prato, Nano Lett., 2006, 6, 1408 CrossRef CAS.
- Q. H. Wang and M. C. Hersam, Nat. Chem., 2009, 1, 206 CrossRef CAS.
- M. Z. Hossain, M. Walsh and M. C. Hersam, J. Am. Chem. Soc., 2010, 132, 15399 CrossRef CAS.
- H. Hu, B. Zhao, M. A. Hamon, K. Kamaras, M. E. Itkis and R. C. Haddon, J. Am. Chem. Soc., 2003, 125, 14893 CrossRef CAS.
- M. S. Strano, C. A. Dyke, M. L. Usrey, P. W. Barone, M. J. Allen, H. Shan, C. Kittrell, R. H. Hauge, J. M. Tour and R. E. Smalley, Science, 2003, 301, 1519 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51 CrossRef CAS.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095 CrossRef CAS.
- A. Jorio, E. H. M. Ferreira, M. V. O. Moutinho, F. Stavale, C. A. Achete and R. B. Capaz, Phys. Status Solidi B, 2010 DOI:10.1002/pssb.201000247.
- C. Mapelli, C. Castiglioni, G. Zerbi and K. Mullen, Phys. Rev. B: Condens. Matter, 1999, 60, 12710 CrossRef CAS.
- C. Castiglioni, M. Tommasini and G. Zerbi, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2425 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS.
- M. S. Strano, C. B. Huffman, V. C. Moore, M. J. O'Connell, E. H. Haroz, J. Hubbard, M. Miller, K. Rialon, C. Kittrell, S. Ramesh, R. H. Hauge and R. E. Smalley, J. Phys. Chem. B, 2003, 107, 6979 CrossRef CAS.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273 CrossRef CAS.
- B. Zhao, M. E. Itkis, S. Niyogi, H. Hu, D. Perea and R. C. Haddon, J. Nanosci. Nanotechnol., 2004, 4, 995 CrossRef CAS.
- B. Zhao, M. E. Itkis, S. Niyogi, H. Hu, J. Zhang and R. C. Haddon, J. Phys. Chem. B, 2004, 108, 8136 CrossRef CAS.
- E. Bekyarova, M. E. Itkis, N. Cabrera, B. Zhao, A. Yu, J. Gao and R. C. Haddon, J. Am. Chem. Soc., 2005, 127, 5990 CrossRef.
- M. J. Calhorda, C. F. Frazao and J. A. M. Simoes, J. Organomet. Chem., 1984, 262, 305 CrossRef CAS.
- W. M. Lamanna, J. Am. Chem. Soc., 1986, 108, 2096 CrossRef CAS.
- P. L. Stanghellini, E. Diana, A. Arrais, A. Rossin and S. F. A. Kettle, Organometallics, 2006, 25, 5024 CrossRef CAS.
- X. Sun, P. Ramesh, M. E. Itkis, E. Bekyarova and R. C. Haddon, J. Phys.: Condens. Matter, 2010, 22, 334216 CrossRef.
- L. H. Liu and M. D. Yan, Nano Lett., 2009, 9, 3375 CrossRef CAS.
- A. R. Pape, K. P. Kaliappan and E. P. Kundig, Chem. Rev., 2000, 100, 2917 CrossRef CAS.
- E. P. Kundig, Topics Organomet. Chem., 2004, 7, 3 Search PubMed.
- H. A. Skinner and J. A. Connor, Pure Appl. Chem., 1985, 57, 79 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR-spectra of the starting chromium reagents and the products, FAB-MS data of the de-complexation reactions, XPS data, absorption spectra, NMR and thermogravimetric analysis data. See DOI: 10.1039/c0sc00634c |
| This journal is © The Royal Society of Chemistry 2011 |