Photosensitised pyrimidine dimerisation in DNA
M. Consuelo
Cuquerella
,
Virginie
Lhiaubet-Vallet
,
Francisco
Bosca
and
Miguel A.
Miranda
*
Instituto de Tecnología Química UPV-CSIC, Universidad Politécnica de Valencia, Avda de los Naranjos s/n, 46022, Valencia, Spain. E-mail: mmiranda@qim.upv.es; Fax: +34 963879444; Tel: +34 963877807
First published on 4th May 2011
Abstract
Triplet-mediated pyrimidine (Pyr) dimerisation is a key process in photochemical damage to DNA. It may occur in the presence of a photosensitiser, provided that a number of requirements are fulfilled, such as favourable intersystem crossing quantum yield and high triplet energy. The attention has been mainly focused on cyclobutane pyrimidine dimers, as they are by far the most relevant Pyr photoproducts obtained by sensitisation. The present perspective deals with the involved chemistry, not only in DNA but also in its simple building blocks. It also includes the photophysical characterisation of the Pyr triplet excited states, as well as a brief discussion of the theoretical aspects.
 Consuelo Cuquerella | M. Consuelo Cuquerella was born in Benigànim (Spain). She obtained her Ph.D. at the Technical University of Valencia working at the Institute of Chemical Technology (UPV-CSIC), under the supervision of Prof. Miguel A. Miranda on DNA damage induced by fluoroquinolones. In June 2004 she moved to the Department of Physics of the University of Liverpool as a post-doctoral Marie Curie fellow. Back to Spain in 2007, she was granted with a Juan de la Cierva contract to work on gold nanoparticles photophysics at the University of Valencia. In 2009, she came back to the Institute of Chemical Technology as a JAE-Doc researcher. |
 Virginie Lhiaubet-Vallet | Virginie Lhiaubet-Vallet was born in La Bachellerie (Aquitaine, France). She graduated in 1997 and obtained her PhD degree in 2001 from the University Paul Sabatier (Toulouse, France), working on DNA damage photoinduced by nonsteroidal anti-inflammatory drugs. She then joined the group of Prof. M. A. Miranda at the Institute of Chemical Technology (UPV-CSIC) as a postdoctoral researcher benefiting from an Individual Marie Curie European Fellowship. Virginie Lhiaubet-Vallet received the Young Investigator Award from the European Society for Photobiology in 2007. Since 2008, she has been a “Ramón y Cajal” Researcher from Spanish National Research Council at the Institute of Chemical Technology. |
 Francisco Boscá | Francisco Boscá, native of Oliva (Spain), completed his PhD in Organic Chemistry at the University of Valencia in 1992. After postdoctoral periods at the University of Illinois (1994) and at the University of Wales (1995), he joined the Institute of Chemical Technology (ITQ) of the Technical University of Valencia as postdoctoral researcher. He is currently a staff scientist (since 1999) at ITQ as member of the Spanish National Research Council (CSIC). His research focus is on photophysical and photochemical properties of different types of photosensitisers looking for possible applications in the development of solar cells and phototherapeutic agents. |
 Miguel A. Miranda | Miguel A. Miranda is Professor of Organic Chemistry at the Technical University of Valencia and Head of the Institute of Chemical Technology. He was Associate Professor at the University of Valencia before accepting his present position in 1990. His research interests are mainly focused on photochemistry and photobiology. Miguel A. Miranda has received the Honda-Fujishima Award of the Japanese Photochemistry Association, the Organic Chemistry Award of the Spanish Royal Society of Chemistry and the Theodor Förster Award of the German Chemical Society and the Bunsen Society of Physical Chemistry. He is currently the President of the European Society for Photobiology. |
1. Introduction
Ultraviolet solar radiation reaching the Earth's surface comprises wavelengths ranging from 290 to 320 nm (UVB) and 320 to 400 nm (UVA). Both UVB and UVA radiations have been demonstrated to induce mutations in DNA that are in the origin of skin cancer. This is a public health problem, aggravated by the increasing use of tanning sunbeds by the general public. Tanning lamps are intended to produce UVA, but they also emit marginally in the UVB. Recently (July 2009) the International Agency for Research of Cancer (IARC) has declared these devices as “carcinogenic to humans”, since they have been proven to increase the risk of skin cancer by 75% when used by people under 30 years old.1,2Although, in principle, longer-wavelength light is less dangerous, it has to be taken into account that defence mechanisms of the human skin towards their deleterious effects are less effective against UVA induced damage.3 Actually, a number of reports have appeared on the promutagenic character of UVA radiation. Thus, studies performed on animals (opossum,4 fish,5 mice6) suggest that it provokes the formation of papillomas, squamous cell carcinomas (SCC) and melanomas; however, the role played by UVA-mediated oxidative damage to DNA in melanoma induction, using xiphophorus fishes as model, has been recently questioned.5,7
While UVB is efficiently absorbed by the nucleobases, causing direct photoreactions of DNA, UVA-induced damage is commonly the result of photosensitisation. Thus, modifications in DNA may occur after light absorption by endogenous or exogenous chromophores present in drugs, cosmetic agents, metabolites, etc.
In this context, UVA-photocarcinogenesis has been mostly related to oxidative stress in early studies. Singlet oxygen production and to a lesser extent hydroxyl radical may be involved in the oxidation of guanine (the nucleobase with the lowest redox potential), giving rise to 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo). However, cyclobutane pyrimidine dimers (CPDs) may also arise from UVA irradiation, and their formation yield is even larger than that of 8-oxo-dGuo in human skin.3,8 This has been assessed by exposure of healthy human volunteers to UVA light and subsequent analysis of CPDs in their urine or skin.9,10
While the promutagenic character of UVA light is established, the mechanism responsible for UVA photoinduced CPDs formation is a matter of discussion. In particular the possibility of direct UVA-photoinduced damage to DNA, in addition to photosensitisation by endogenous or exogenous agents, is still controversial.3,8,11–17 It has been claimed that CPDs may be formed after UVA irradiation of isolated or cellular DNA in the absence of a photosensitiser. However DNA hardly absorbs UVA, as required for a molecule to react (first law of photochemistry, Grotthus–Draper law). As an alternative to direct DNA excitation, the presence of unknown chromophores or the insufficient purity of the UVA sources used in the experiments has been considered. Moreover, cellular DNA irradiations produce less CPDs in comparison with isolated DNA. As it is difficult to estimate the amount of light absorbed by DNA under these conditions, a contribution by endogenous photosensitisers (i. e.porphyrins, flavins, steroids, quinones) cannot be safely ruled out.
The aim of the present perspective article is to present the case of UVA-photosensitised damage to DNA, with special emphasis on the molecular mechanisms involved in the formation of CPD lesions. A better understanding of these processes should contribute to minimise the photobiological risk.
Among the exogenous agents reported to be photogenotoxic, phototumorigenic and photocarcinogenic in vivo, psoralens (used in the PUVA treatment of psoriasis)18–21 and more recently fluoroquinolones (FQs), have received special attention (Fig. 1). The latter are widely used, broad spectrum antibacterial drugs. They are known to induce UVA-mediated oxidatively damaged DNA,22–25 and their phototumorigenic potential has been proven in mice.26–29 Irradiation of albino Swiss and skh-1 hairless mice with UVA light, varying the time of exposure and the drug doses, has established that fleroxacin (FLX) and lomefloxacin (LFX) are more potent phototumorigenic agents than 8-methoxypsoralen (8-MOP). Development of SCCs after the intake of FLX or LFX, together with other lesions such as benign papillomas, solar keratoses or kerato-acanthomas has also been observed in the rodents. Likewise, ofloxacin (OFX), ciprofloxacin (CPX) and the related compound nalidixic acid (NA) have also been found to enhance the development of skin tumours.
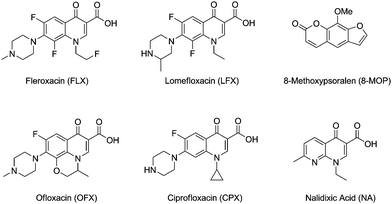 | ||
| Fig. 1 Exogenous photosensitisers acting as photocarcinogens. | ||
In vivo studies on xeroderma pigmentosum mice have also revealed LFX as a photocarcinogenic agent. These mice present an inefficient nucleoside excision repair activity for the enzymatic removal of CPDs29 while conserving the capability to repair oxidatively damaged DNA. Exposure of mice to low UVA doses insufficient to provoke severe phototoxic reactions, leads to a large number of SCCs after only 5 weeks; by contrast, control animals require 23 weeks to show similar effects.
2. Photosensitised formation of cyclobutane pyrimidine dimers
Photosensitised CPDs formation takes place through a formal [2 + 2] cycloaddition between the C5–C6 double bonds of two pyrimidines (Pyr). Thus, a photosensitiser (Phs) is excited upon light absorption and then transfers its energy to a pyrimidine base, giving rise to thymine or cytosine excited states (Thy* or Cyt*). These states are able to react with ground state Thy or Cyt leading to the final products (Schemes 1 and 2).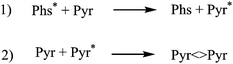 | ||
| Scheme 1 Key processes involved in photosensitised Pyr dimerisation. | ||
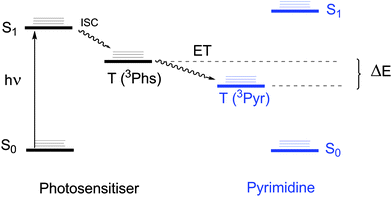 | ||
| Scheme 2 Interconversion between the excited states involved in photosensitised Pyr dimerisation. | ||
In general, sensitised photocycloadditions are known to proceed through a triplet–triplet energy transfer (TTET) process. As a consequence, a number of requirements should be fulfilled by the Phs of choice: i) to absorb light at longer wavelengths than Pyr, thus allowing for selective excitation, ii) to have a triplet energy above that of Pyr, as requested for thermodynamically favoured process (Scheme 2), iii) to be chemically inert under the reaction conditions, avoiding formation of byproducts and consumption of the Phs, iv) to have a good intersystem crossing quantum yield (ϕISC) and a long triplet lifetime (τT), in order to increase the probability of energy transfer to an acceptor, and v) to be close enough to the Pyr unit, thereby facilitating collision.
2.1 Efficiency of photosensitised pyrimidine dimerisation
In a TTET process, the energy transfer rate constant (kET) between the Phs (donor) and the Pyr (acceptor) depends on the energy gap (ΔE), as shown by Sandros' equation: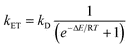 | (1) |
In this context, a favourable value of ΔE is directly related to CPDs formation and therefore to the phototumorigenic capability of endo- or exogenous agents acting as Phs. Thus, it is of paramount importance to establish Pyr triplet excited state energies (3Thy*, 3Cyt*) to anticipate the potential of a compound to act as Phs. Consequently, an effort has been made in this sense, with special attention to 3Thy in DNA, where π-stacking and base pairing can have a marked influence on the triplet excited state properties.
In addition to ΔE and temperature18 (as inferred from eqn (1)), photoproducts formation and distribution are influenced by parameters such as the nature of the lowest lying triplet state of the Phs,31 the solvent,32 the concentration of Pyr used, etc.
The importance of Pyr concentration deserves a special comment. According to Scheme 1, ground state Pyr quenches both 3Phs* and 3Pyr*, so the reaction rates should increase with increasing Pyr concentration. In practical terms, photodimerisation quantum yields (ϕD) are reproducible only upon complete quenching of the involved triplet excited states. Accordingly, only a limited number of ϕD values are available (Table 1), due to the experimental difficulties to cover all the above requirements; they range between 10−2 and 10−5. The values are consistent with the upper limit established by measurements performed using acetonitrile (0.02), where formation of cyclobutane thymine dimers (Thy<>Thy) is assumed to occur almost exclusively through the triplet excited state.33
| Substrate | Concentration | Photosensitiser | ϕ D |
|---|---|---|---|
| a In aqueous solution. b In ethyl acetate, toluene, methanol or acetonitrile solutions. c BP, benzophenone; PABA, para-aminobenzoic acid; DMT, dimethylthymine. | |||
| Thy | 1 × 10−2 Ma | Acetone 37 | 0.0042 |
| Acetophenone 37 | 0.0016 | ||
| BP37 | 0.011 | ||
| PABA 38 | 0.0007 | ||
| DMT c | 0.1 Mb | BP39 | 0.023 |
| 0.05 Mb | Acetophenone 39 | 0.017 | |
| 0.1 Mb | 0.061 | ||
| 0.2 Mb | 0.031 | ||
| Supercoiled DNA | 18.85 μM in base pairs | Tiaprofenic acid 40 | 0.00001 |
| Ketoprofen 40 | 0.0002 | ||
| Acetophenone 40 | 0.006 | ||
| Phage T4 | — | Cationic acetophenone41 | 0.03 |
2.2 Pyrimidine photoproducts in DNA building blocks
In principle, Thy<>Thy, Cyt<>Thy, and Cyt<>Cyt dimers can be formed by photosensitisation of solutions containing the appropriate monomers. Different regio-and diastereoisomers may be obtained in solution (see Fig. 2).34 It is worth noting that the cis-anti and trans-syn isomers exist as enantiomeric pairs in Thy and Cyt and as diastereomeric pairs in thymidine (Thd) and 2′-deoxycytidine (dCyd). Thus a total of 6 isolable diastereomeric homodimers can be obtained from the nucleosides, as compared with 4 in the case of the free bases.34,35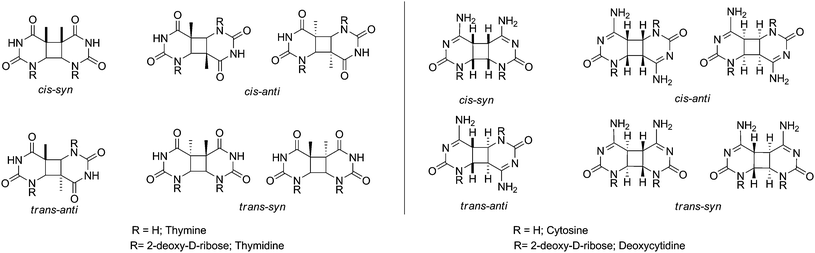 | ||
| Fig. 2 Structures of all possible homodimers formed after UVA-photosensitised irradiation of Pyr. | ||
Although crossed cycloadditions are, in principle, possible to form Thy<>Cyt heterodimers, photosensitised formation of these CPDs has not been described. In this context, Thy dimers have received special attention, since they are the most abundant dimers formed in DNA.6,8,15,36
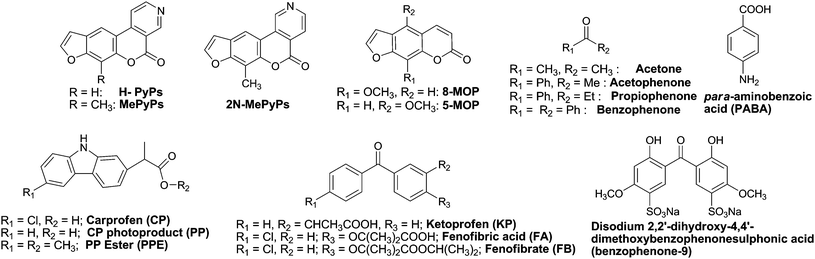 | ||
| Fig. 3 Photosensitisers for Pyr dimerisation. | ||
When pyridopsoralens with different triplet energies (ET), namely, pyrido[3,4-c]psoralen (H-PyPs, ET = 290.4 kJ mol−1), 7-methylpyrido[3,4-c]psoralen (MePyPs, ET = 288.5 kJ mol−1) and 7-methylpyrido[4,3-c]psoralen (2N-MePyPs, ET = 281.7 kJ mol−1) are irradiated in thin films in the presence of Thy, Thy<>Thy dimers are obtained with the cis-syn isomer as the most abundant one.18 Products yields correlate with the PyPs triplet energies, as expected from Sandros' equation. Furthermore, parallel irradiations with the related compounds 5-methoxypsoralen (5-MOP, ET = 268.2 kJ mol−1) and 8-MOP (ET = 260.5 kJ mol−1) show very little if any photosensitised dimer formation.
A clear example of the temperature effect is provided by an experiment performed with H-PyPs. Photoinduced Thy<>Thy formation at 77 K is two orders of magnitude less efficient than at 300 K. Photosensitisers with ET lower than that of 3Pyr*18,42,43 can still work upon thermal population of the 3Phs* upper vibrational states. Even if formed in low yields, the resulting CPDs would be of biological significance.18,31
In this context, distribution of the photodimers mixture can be influenced by the polarity of the solvent. Thus, benzophenone (BP) photosensitisation of 1,3-dimethylthymine (DMT) leads to the cis-syn isomer as the major photoproduct in polar solvents (∼72%, CH3CN and CH3OH) while in non-polar solvents the cis-anti one predominates (∼50%, benzene).32,39
Ketones have often been used to photosensitise CPDs formation taking advantage of their high ϕISC (nearly 1).32,37,44–49 Irradiation of aqueous solutions of Thy (or Thd) in the presence of acetone, propiophenone, acetophenone or benzophenone gives rise to a mixture of isomers (Fig. 2), with certain prevalence of the trans-anti diastereomers.44,46–48
Interestingly, ketones can also mediate oxidation of Pyr bases given their ability to participate in hydrogen abstraction or electron transfer processes50 (Scheme 3). Nonetheless, in the case of dinucleotides such as TpT48 energy transfer prevails (ca. 94%) over BP-photosensitised oxidation.
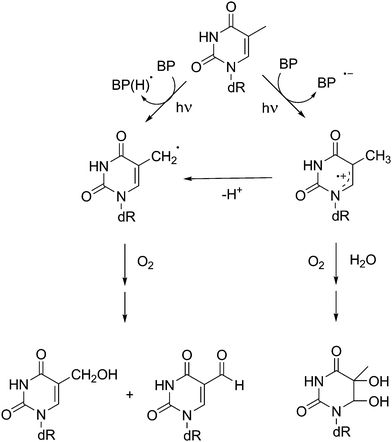 | ||
| Scheme 3 Mechanistic pathways involved in the photosensitised oxidation of thymidine by benzophenone. | ||
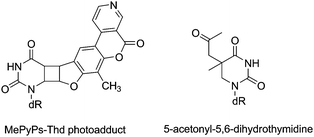 | ||
| Fig. 4 Structures of byproducts obtained upon photosensitisation of Thd with PyPs (left) or acetone (right). | ||
For example, PyPs possess a double bond in their furan moiety prone to react through a [2 + 2] cross-photocycloaddition with the C5–C6 double bond of Pyr. Actually, when CPDs formation is photosensitised by PyPs, this type of photoaddition is indeed observed.19 Likewise in the acetone photosensitised reaction of Thd, an acetonyl derivative has been isolated as a side product.34
Furthermore, carbonyl compounds may in principle react with alkenes to form oxetane derivatives through a [2 + 2] photocycloaddition (Paterno–Büchi reaction). This process is favoured when i) the triplet energy of the alkene is comparable to (or higher than) that of the carbonyl compound and ii) the lowest lying carbonyl triplet state is of nπ* nature. As a consequence, ketones with relatively low triplet energy may lead to oxetane derivatives of Pyr, in addition to CPDs.51–53 This is the case of BP:31,44 its triplet excited state, which presents a nπ* configuration, has an ET level below that of acetone or acetophenone. As a matter of fact, a Paterno–Büchi reaction giving rise to oxetanes is favoured versusTTET (Scheme 4). Only when Thd is present at high concentrations CPDs are obtained.31,50 Similar observations have been reported for NSAIDs containing the BP chromophore, such as ketoprofen (KP) and its derivatives.51,52,54,55 Similarly, a photocycloaddition product identified as an oxetane has been described after irradiation of cytosine in acetone–aqueous (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solutions.53
:1) solutions.53
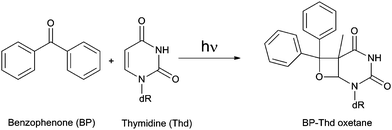 | ||
| Scheme 4 Paterno–Büchi photoreaction between BP and Thd. | ||
2.3 Oligonucleotides photosensitisation
Photosensitised CPDs formation has been observed in oligonucleotides and single stranded DNA (ss-DNA). Here, Pyr dimerisation occurs through adjacent Pyr on the same strand, inducing a distortion in the structure. Among the possible cyclobutane dimers, 5′-Thy<>Thy-3′, 5′-Cyt<>Thy-3′ and 5′-Thy<>Cyt-3′ are preferentially promoted. However, 5′-Cyt<>Cyt-3′ may be obtained in lower yields.6 Since Cyt is the DNA base with the highest triplet energy, a reduced number of photosensitisers can be involved in a thermodynamically favourable TTET. Furthermore, if a fraction of Cyt reaches the triplet excited state, efficient deactivation by energy transfer to the other bases should be expected.36In addition, Thy<>Thy predominate over Thy<>Cyt dimers in oligonucleotides and DNA.56 For instance, acetophenone-mediated photodimerisation of thymidylyl-(3′-5′)-thymidine (TpT, Scheme 5) occurs five to six times faster than that of thymidylyl-(3′-5′)-deoxycytidine (TpdC).57 The energy gap values in terms of Sandros' equation (eqn (1)), together with the relative reactivity of 3Thy towards Cyt or Thy, would explain the different photodimerisation rates.
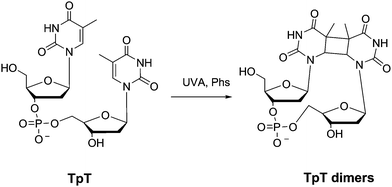 | ||
| Scheme 5 Photosensitised thymidylyl-(3′-5′)-thymidine (TpT) dimerisation. | ||
Orientation restrictions imposed by the sugar–phosphodiester backbone prevent formation of the anti forms and favour the cis-syn arrangement;19,35,48,55–59 nonetheless, trans-syn dimers are also observed in ss-DNA or oligonucleotides owing to their flexible structure.56,60 This is the case of dCpT, TpdC and TpT, which produce mainly cis-syn and trans-syn diastereomers48,61 in proportions that may range from 7:1 (TpT) to 3:1 (dCpT) or 1:1 (TpdC). Similar results are obtained when two Thy units are kept in close proximity through a polymethylene linker.62 The largest Thy<>Thy yield is obtained in the case of Thy–(CH2)3–Thy, likely because the angle between the two Thy approaches that in DNA. Only the intramolecular photodimerisation of some N-acetylated dinucleotides gives rise exclusively to the trans-syn configuration, presumably due to steric hindrance.63
The influence of the Thy–Thy distance has also been evaluated by comparing the reaction rates and Thy<>Thy formation yields of poly(T) and depurinated poly(dA–T). In both cases, Thy<>Thy dimers are formed, albeit the reaction rate is slowed down in the latter due to the poorer π-stacking and longer base-to-base distance. Conformation has a pronounced effect on CPDs formation by governing the extent of stacking between the bases. This is further supported by the fact that dimerisation efficiency is reduced after denaturation by addition of a suitable solvent like ethanol.36,64,65
The outcome of dimerisation does not only depend on the conformation but also on the nucleobase sequence.66 The frequency of CPDs lesions increases when a Pyr is located in the 5′ side of two consecutive Thy.56,67–70 Studies performed by means of 32P radiolabelling and subsequent electrophoresis combined with specific DNA repair enzymes, have shown that photosensitised Pyr<>Pyr formation in a 25-mer 5′-TGA GCG TTA GTT TAA GTC GGC TATC-3′ by ketonic drugs occurs more frequently in TTT fragments.
2.4 DNA photosensitisation
While UVA-photosensitisation of DNA gives rise exclusively to CPDs, direct UVB irradiation also produces pyrimidine (6–4) pyrimidone photoproducts.16 This strongly suggests the involvement of two different mechanisms.As in oligonucleotides, DNA photosensitisation produces Thy<>Thy, 5′-Cyt<>Thy-3′ and 5′-Thy<>Cyt-3′, together with small amounts of Cyt<>Cyt in adjacent pyrimidines on the same strand, with an overwhelming predominance of cis-syn Thy<>Thy. Analysis of these lesions is often performed by radiolabelling and subsequent electrophoresis.25,71,72 Single cell electrophoresis (comet assay) has been successfully used to reveal cellular DNA damage, as fragmented DNA moves faster through the agarose gel, forming a tail.73,74 Both methods can be combined with specific repair enzymes to reveal the type of damage produced.
Relative Pyr<>Pyr formation yields are listed in Table 2. For example, irradiation of DNA using BP or acetophenone as Phs does not produce detectable amounts of Cyt<>Cyt. Only acetone photosensitisation leads to Cyt homodimers,8,56,75,76 presumably due to the higher ET. In general, 5′-Cyt<>Thy-3′ and 5′-Thy<>Cyt-3′ photosensitisation is inefficient as compared to Thy<>Thy, although in ct-DNA and coliphage M13 considerable amounts of heterodimers are obtained. For comparison, in direct DNA photolysis the relative formation yields are 1:0.8:0.2 (TT:CT:CC).36
| BPa | Acetophenone a,b,c,d,e | Acetone e,f | ||||||
|---|---|---|---|---|---|---|---|---|
| a | a | b | c | d | e | e | f | |
| a Calf-thymus DNA.8 b Native DNA.64 c Denatured DNA.64 d E. coli DNA.41 e Phage T7.77 f Coliphage M13 mp2.56 g n.d. = Not determined. For each experiment, the amount of Thy<>Thy has been set as the unity for comparison. | ||||||||
| Thy<>Thy | 0.2 | 1 | 1 | 1 | 1 | 0.65 | 1 | 1 |
| Thy<>Cyt | 0.046 | 0.24 | 0.05 | 0.19 | 0.03 | n.dg | 0.12 | 0.20 |
| Cyt<>Thy | 0.05 | 0.23 | 0.36 | |||||
| Cyt<>Cyt | n.d.g | n.d.g | n.d.g | n.d.g | <0.003 | n.d.g | 0.05 | 0.08 |
Formation of CPDs in isolated and cellular DNA can be photosensitised not only by ketones,58,76 but also by PyPs,19,78 NSAIDs,40,54,68,79 FQs,23,25,29,71–74,80,81amino acids and derivatives59,82 or cosmetic agents83,84 (Fig. 5). Studies performed on Thy dimerisation by the FQ family have played a key role in determining the triplet energy of Thy in DNA at ca 267 kJ mol−1. Dimers formation is mediated by ENX and NFX, while it is not by the N(4′)-acetyl NFX derivative (ANFX) or OFX.23,25,71,72 Interestingly, for ENX, LFX and NFX the efficiency of CPDs formation has been found to be different in isolated and cellular DNA.
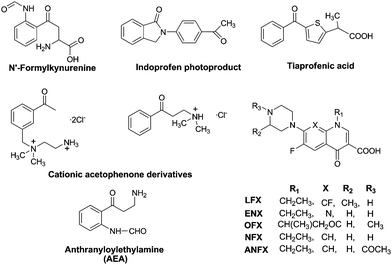 | ||
| Fig. 5 Photosensitisers for Pyr<>Pyr formation in DNA. | ||
As in oligonucleotides, Thy<>Thy formation in DNA is sequence-dependent.19,56,69,78 Pyridopsoralens H-PyPs, MePyPs and 2N-MePyPs react specially at TTTTA and TTAAT fragments, provoking 40% and 55% of Thy<>Thy photolesions, respectively. Moreover, CPDs formation is not detected in a GC environment or at CC sites.
In addition to neighbouring effects, sequence dependence may be the result of selective formation of Phs-DNA complexes in specific DNA locations. Thus, complexation to DNA can place the Phs and the Pyr units in close proximity favouring TTET processes. As an example, 4′,5′-dihydro-7-methylpyrido[3,4-c]psoralen, a modified PyPs, binds to DNA close to 5′-TA-3′ sites.78 If the binding is disrupted (i.e. by varying the ionic strength), Thy dimers formation is negligible.82 An additional example is provided by two cationic derivatives: β-dimethylaminopropiophenone hydrochloride and N-(m-acetylbenzyl)-N-(2-aminoethyl) ammonium dichloride. The charged Phs are brought close to DNA by ionic interactions, which is reflected in an increased photosensitisation capability with respect to acetophenone.41
3. Spore photoproducts
Another interesting dimeric pyrimidine lesion corresponds to the most abundant UV photoproduct in bacterial spores.85,86 Indeed, 5-thyminyl-5,6-dihydrothymine adduct, the so called spore photoproduct (Fig. 6, SP), is formally obtained by linking the allylic carbon to the C5 position of a neighbouring Thy, with saturation of the C5–C6 double bond. Four isomeric forms of SP may be formed within DNA since a chiral center is generated at C5a carbon. Furthermore, the allylic carbon can be linked to adjacent thymines located either at the 3′- or at the 5′-end. Interestingly, DNA double helix structure induces a highly stereospecific formation of SP photoproducts (Fig. 6);87 their stucture has been recently assigned by 2D NMR studies combined with DFT calculations. Thus, the natural SP results from addition of the thymine methyl group located on the 3′-end to the thymine C5 carbon located in the 5′-end, giving rise to a new chiral center with R absolute configuration.87 Moreover, SP is only obtained as a Thy homodimer and has been detected both as intrastrand and interstrand lesion.88,89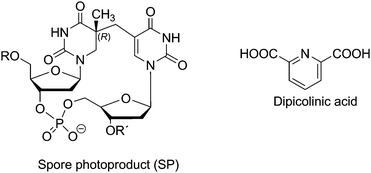 | ||
| Fig. 6 Structures of SP photoproduct and of dipicolinic acid. | ||
Indeed, SP is a quite peculiar bipyrimidine photoproduct, whose formation has been related to three important factors:85,89,90 i) the low hydration level in spore core, ii) the binding of α/β type small, acid soluble protein (SASP), which converts DNA from B-like to A-like conformation, and iii) the presence of dipicolinic acid (pyridine-2,6-dicarboxylic acid DPA, Fig. 6) in the spore core (up to 10% of dry weight). The photosensitising properties of this endogenous compound have been first proposed on the basis of the decrease of SP formation yield in spore strains lacking DPA.88,91 This hypothesis has been further supported by studies in less complex media like isolated DNA, TpT or Thd.49,87–89,92,93 In this context, UVC irradiation of DNA dry films in the presence of DPA has revealed the increase of SP, Thy<>Thy, and to a lesser Thy<>Cyt and Cyt<>Thy relative yields,85,88,91 whereas Cyt<>Cyt and (6–4) photoproducts yields remain almost unchanged. These data are in agreement with the role of DPA as triplet photosensitiser, acting as donor in TTET processes. Further pieces of evidence supporting the feasibility of such a process have been provided by the results of UVA-irradiation of Thd dry films in the presence of BP or three pyridopsoralen derivatives (H-PyPs, MePyPs and 2N-MePyPS, Fig. 3).19,49 In these experiments, the six diastereoisomers of Thd<>Thd and the 5R*/5S* diastereoisomers of SP have been detected; their yields have been shown to depend on the nature of the photosensitiser.19 As expected for triplet energy donors, their efficiency can be related to their excited state level, with H-PyPs being the most efficient, followed by MePyPs and 2N-MePyPs. Accordingly, no formation of Thd<>Thd and SP has been observed for the donors with lower triplet energy like 3-carbethoxypsoralen, 5-methoxypsoralen and 8-MOP.19
Finally, it is noteworthy that in spite of the interesting SP photochemistry, the involved mechanism has not been investigated.85,94 The reaction may occur by coupling of the 5-thyminyl /5,6-dihydrothymin-5-yl radical pair generated as a result of H-abstraction from a ground state by a triplet excited Thy (Scheme 6). Alternatively, a concerted mechanism involving the methyl group of one Thy and the double bond of the second Thy has been proposed. Either the shorter lifetime of radical pairs (as compared with the free radicals generated by radiation)95,96 or the concerted nature of the process would account for the observed stereoselectivity.
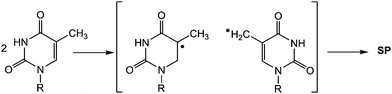 | ||
| Scheme 6 Mechanism postulated for SP formation. | ||
4. Theoretical calculations
In spite of their importance in DNA damage formation, the Pyr triplet excited states are only now starting to be analysed by theoretical studies.97–108 First principles calculation methods converge on the ππ* nature of the lowest triplet state but not on their energy values, which have been reported over a large range (Table 3) depending on the method used for calculation and geometry optimisation. So far, the highest level of approach has been CASPT2 based on CASSCF wave functions. However, because of the difficulties to apply this methodology for large molecules, a number of calculations have been performed at the density functional theory (DFT) level or by using the coupled cluster (CC) model.| Vertical | Adiabatic | |
|---|---|---|
| a Calculated using DFT. b Calculated using CCSD. c Calculated using CC2. d Calculated using CASPT2//CASSCF. | ||
| Thy | 272.1–275.0 (2.82–2.85)a,97 | |
| 336.7–337.7 (3.49–3.50)a,97 | 288.5–296.2 (2.99–3.07)b,97 | |
| 366.6–377.3 (3.80–3.91)b,97 | 299.1–304.9 (3.10–3.16)c,98 | |
| 358.9–372.4 (3.72–3.86)c,98 | 285.6 (2.96)a,98 | |
| 341.6 (3.54)a,98 | 296.2 (3.07)c,100 | |
| 373.4 (3.87)c,100 | ||
| 376.3 (3.90)c,100hyd, | ||
| 382.1 (3.96)c,100aq | ||
| 346.4 (3.59)d,99 | 276.9 (2.87)d,99 | |
| 368.6 (3.82)c,107 | ||
| Thy–Thy | 274.6–275.5 (2.84–2.85)a,102 | |
| 227.7 (2.36)d,103 | ||
| Cyt | 347.3–350.2 (3.60–3.63)a,97 | 293.3–298.1 (3.04–3.09)a,97 |
| 361.8–383 (3.75–3.97)b,97 | 302.0–311.6 (3.13–3.23)b,a | |
| 352.2 (3.65)d,101 | 293.3 (3.04)d,101 | |
| 330.0–352.2 (3.42–3.65)a,105 | 284.6–298.1 (2.95–3.09)a,105 | |
| 361.8–383.0 (3.75–3.97)b,105 | 302.0–311.6 (3.13–3.23)b,105 | |
| 340.6 (3.53)d,106 | 287.5 (2.98)d,106 | |
| 374.4 (3.88)c,107 | ||
| Cyt–Cyt | 260.5 (2.70)d,103 | |
| Ura | ||
| 284.6–383.0 (2.95–3.97)a,97 | ||
| 287.5–419.7 (2.98–4.35)b,97 | ||
| 370.5–383.0 (3.84–3.97)c,98 | 312.6–319.4 (3.24–3.31)c,98 | |
| 367.7 (3.78)a,98 | 302.0 (3.13)a,98 | |
| 384.0 (3.98)c,100 | 309.7 (3.21)c,100 | |
| 387.9 (4.02)c,100hyd | ||
| 395.6 (4.10)c,100aq | ||
| 366.6 (3.80)d,108 | 303.9 (3.15)d,108 | |
| 379.2 (3.93)c,107 | ||
| Ura – Ura | 238.3 (2.47)d,103 |
In this context, the vertical excitation energy for Thy at the ground state geometry is situated between 336.7 and 382.1 kJ mol−1 (3.49 eV and 3.96 eV), while adiabatic excitation energy values range from 272.1 to 304.9 kJ mol−1 (from 2.82 to 3.16 eV). Similar discrepancies have been reported for Cyt and Ura (Table 3). Nevertheless, the general trend extracted from the calculated values is in agreement with the experimental data, i.e. the nucleobase with the lowest triplet excited state energy is Thy, while the highest triplet manifold corresponds to Cyt. Recently, the energetic gap between 3Thy* and 3Ura* has been rationalised in terms of the influence of C5-methylation. This substitution induces an up-shift of the π HOMO while the acceptor π* LUMO is almost unaffected, thus resulting in a red shifted π → π* transition.100
Computational studies are generally performed in vacuo i.e. without taking into account interaction between the target molecules and solvent. However, it is well established that such interactions may have a marked influence on the excited state energy. In this context, the combined effect of hydration (energies denoted as hyd, Table 3) and solvent polarity, used to mimic aqueous environment (denoted aq in Table 3) has only revealed a slight destabilisation of the lowest 3ππ* triplet state of Ura and Thy.100,109 Concerning the photochemical reactivity of Pyr, theoretical calculations have mainly focused on the formation of cyclobutane dimers in a concerted process from the singlet excited state.110–114 However, as stated above, in the case of photosensitised Pyr<>Pyr formation, [2 + 2] photocycloaddition occurs from the triplet manifold. In spite of its importance, up to now this reaction pathway has only been considered by two research teams.102–104 In a first approach, calculations at the TD-DFT level have shown that triplet mediated photocycloaddition of Thy proceeds through an initial C6–C6′ bond formation, leading to a biradical intermediate that subsequently crosslinks to the singlet surface, giving rise to Thy<>Thy.102 More recently, Pyr<>Pyr formation has been computed by means of high level quantum chemical CASPT2//CASSCF calculations.103,104 For a better approach to the nucleobase properties in DNA, the triplet minima have been calculated for excimer arrangements formed by the parallel stacking of the bases (Scheme 7). In this context, a stabilisation relative to the isolated bases (denoted 3Pyr*+Pyr in Scheme 7) is observed, giving rise to adiabatic energy values of 2.36, 2.47, 2.70 eV for Thy, Ura, and Cyt, respectively. This triplet excimer connects without any energy barrier with a stepwise intermediate 3(SWI), which exhibits a biradical character. Indeed, a covalent bond is formed between the C6 and C6′ carbons, with the unpaired electrons and spin density located on the two other ethylenic carbons (C5 and C5′). At this stage, the calculated C6–C6′ bond lengths are 1.669, 1.660 and 1.664 Å for Cyt, Ura, and Thy; by contrast, the interatomic C5–C5′ distance is elongated (about 2.8 Å). Finally, 3(SWI) corresponds to a singlet–triplet crossing (T1/S0)x structure leading to Pyr<>Pyr ground state.
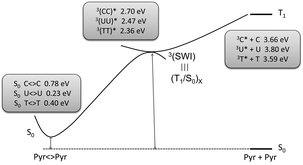 | ||
| Scheme 7 Triplet mediated formation of Pyr<>Pyr. Adapted from Serrano et al.103 | ||
Thus, the efficiency of Pyr<>Pyr photosensitisation depends on two factors. The first one is the effectiveness of the TTET process from the 3Phs*, which is related with the triplet state energy of the nucleobase (3Cyt*>3Ura*>3Thy*); this agrees with the experimental predominance of Thy<>Thy dimers vs. other Pyr combinations. The second factor deals with the efficiency of the intersystem crossing process toward the ground state of the photoproduct (T1/S0)x.
5. Photophysics
5.1 Emission
Thymine and its derivatives exhibit a pH-dependent phosphorescence emission. At 77 K under neutral conditions, no signal is observed for diluted aqueous solutions.115–117 Conversely, at a pH higher than the pKa of Thy (ca. 9.6),118 the nucleobase in its anionic form emits at 445 nm with a decay time of 0.4–0.5 s.115,119,120 Parallel experiments have reported that at neutral pH and high concentrations (i. e. 10−3–10−2 M), formation of aggregates gives rise to a phosphorescence emission at 470 nm, with a lifetime of 0.2 s (Table 4).42,118 A similar spectrum has been detected by photosensitisation experiments using acetone to populate the Thd monophosphate (TMP) triplet excited state by an energy transfer mechanism.42,120 Emission of Cyt and its derivatives has been studied to a lesser extent;43,120–125 the obtained data are listed in Table 4.| λ max (nm) | τ (s) | ϕ em | |
|---|---|---|---|
| a 2-Methyltetrahydrofuran. b In H2O pH 7. c In CH3CH2OH. d 0.25% glucose in 0.1M sodium acetate-H2O. e Isopropanol/isopentane. f H2O/propylene glycol 1/1. g H2O. h CH3OH–H2O 1/9 pH 7. | |||
| Thy | 470118,127 | 0.543 | <0.008122 |
| 460a,129 | 0.075a,129 | 0.00643 | |
| 0.018–0.015a,129 | |||
| Thd | 470b,127 | 0.21b,127 | <0.015122 |
| 460a,129 | ≤0.5c,122 | 0.00643 | |
| 450d,123+ | 0.6d,123 | 0.038–0.042a,129 | |
| TMP | 44042,120 | 0.342,120 | 0.00843 |
| ≤0.4122 | ≤0.01122 | ||
| dApT (or TpdA) | 440120 | 0.021 (0.009)120 | |
| polydAT | 450126 | 0.342,115,119,126,128 | ≥0.004119 |
| 448119 | 0.342,115,119,126 | ||
| DNA | 450126 | ||
| 448119 | 0.3g,119 | 0.002115,119 | |
| 450d,123 | 0.5d,123 | ≤0.02122 | |
| Cyt | 430e,123 | 0.8c,122 | 0.006122 |
| 0.6e,123 | |||
| dCyd | 435b,127 | 0.6b,118 | 0.009122 |
| 430f,b,121 | 0.66c,122 | ||
| 410d,123 | 0.6d,123 | ||
| dCMP | 410h,120,124 | 0.4122 | 0.015122 |
| 0.34120,125 | 0.01120 | ||
| dCpdC | 0.01120 | ||
| polyC | 420d,123 | 0.7119 | 0.02122 |
| 0.6122 |
Thus, as shown in Fig. 7, Thy and Cyt are the nucleobases with the lowest and the highest triplet excited state energies, respectively.
 | ||
| Fig. 7 Energies of DNA bases determined at 77 K in ethylene glycol/H2O.42 | ||
An intriguing result is the reported phosphorescence emission of native DNA. Indeed, by considering the higher phosphorescence quantum yield of purines, an overall emission closely related to these bases could be expected. However, the DNA spectrum does not exhibit the well-structured band characteristic of purines, and the obtained quantum yield (Table 4) is 1 order of magnitude lower than that of adenosine monophosphate (AMP) or guanosine monophosphate (GMP). Hence, it has been proposed that DNA emission arises from the triplet level of Thy residues. Accordingly, a more intense phosphorescence has been monitored for DNA with higher adenine (Ade) + Thy contents.42,126 Different hypotheses have been postulated to explain the nature of the emissive residues. On the basis of the lack of phosphorescence found for isolated Thy, emission was initially attributed to that of Thy anion formed by transfer of the thymine N3 proton across the Watson–Crick base pairing to the N1 nitrogen of adenine.115,119 Nevertheless, this hypothesis has been contradicted by the phosphorescence emission of Thy aggregates, which closely resembles that of DNA. This assignment has been further supported by the Thy-like emission of 1,3-dimethylthymine in the presence of Ade, where the proton transfer is not possible.118,127 A similar conclusion has been drawn from single stranded DNA studies.42 Different explanations have been provided to account for the fact that Thy is the only emitting residue in DNA. The first one is related to its relatively low triplet state energy. It has been reported that, irrespective of the excited chromophore (Ade, Gua, Thy or Cyt), the only emission observed in solutions containing mixtures of the nucleobases is that of 3Thy*.118,120,127 This is consistent with energy transfer from the higher lying triplet excited states of Cyt, Ade or Gua to Thy in solution. Accordingly, a thymine emission has also been obtained for the dinucleotides dApT and TpdA but also for polydAT.42,115,119,120,126,128 A similar deactivation channel towards 3Thy* has been postulated to explain the case of the whole DNA biomacromolecule.118,127
On the other hand, for the Gua–Cyt base pair, proton transfer at the singlet level can also be in the origin of their lack of emission.118,119 Expectedly, DNA structure integrity is an important parameter. Temperature, solvent or pH conditions leading to denaturation of the double helix result in the typical blue shifted, more structured and longer lived phosphorescence emission of purines.119,126
5.2 Laser flash photolysis studies
Table 5 shows a selection of key photophysical parameters (ISC quantum yield, as well as rate constants for unimolecular decay and self quenching, k0), determined for the triplet excited state of the Thy chromophore in the free base and in some derivatives. It also includes the corresponding molar absorption coefficients of the T–T absorption (εT), which have been obtained applying an energy transfer method, with retinol as acceptor.130 The data indicate that formation of the triplet excited states upon direct UV-irradiation occurs actually in all cases. In spite of its biological relevance, the efficiency of this process is low, particularly in aqueous medium.
| Solvent | ε T (M−1 cm−1) at 370 nm141,143 | ϕ ISC /10−2141,143 | k 0 (s−1)/105136,143 | k S (M−1 s−1)/108136,143 | |
|---|---|---|---|---|---|
| a Rate constant given in s−1. | |||||
| Thy | CH3CN | 2700 | 6.0 | 0.7–2.2 | 5.3–7.0 |
| H2O | 3500 | 0.6 | 0.2 | 7.9 | |
| Thd | CH3CN | 3600 | 6.9 | ||
| H2O | 3600 | 1.4 | 0.4 | 1.0–1.9 | |
| TMP | CH3CH2OH | 4000 | 5.5 | 2.0 | |
| H2O | 3500 | 0.8–1.5 | 0.4 | 0.2 | |
| (T)20 | H2O | 2700 | 2.8 | 100a |
As stated above, triplet quenching by the ground states is associated with Pyr<>Pyr dimerisation. Its rate constant can be determined according to:
| kobs = k0 + kS[S0] | (2) |
By contrast with Thy derivatives, the triplet excited states of Cyt, dCyd and dCyd monophosphate (dCMP) have not been observed upon direct photolysis; this can be atributed to the low ISC quantum yields and molar absorption coefficients.133
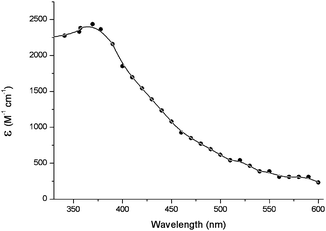 | ||
| Fig. 8 Transient absorption spectrum of triplet excited of TMP, obtained by energy transfer from acetone in deaerated aqueous solutions.134 | ||
After excitation of the photosensitiser, a number of processes may occur. The initial step is formation of the Phs first singlet excited state (1Phs*). At this point, several pathways can compete: fluorescence and internal conversion lead back to Phs, while intersystem crossing affords the triplet excited state (3Phs*). An ideal photosensitiser should have an ISC quantum yield close to 1.
The Phs triplet energy relative to that of the Pyr derivative is a key point to predict whether energy transfer can proceed. Thus, for irreversible energy transfer, the triplet energy of the donor must be at least 12 kJ mol−1 higher than that of the acceptor.135
In this context, the interaction between a variety of ketone triplets and mononucleotides has been studied as a function of the relative energies of the Phs–nucleotide pair. While ketones with ET higher than 305 kJ mol−1 (acetone, acetophenone, propiophenone and 1-indanone) sensitise the generation of a transient absorption corresponding to 3TMP* in laser flash photolysis (LFP), those with ET < 305 kJ mol−1 do not exhibit any triplet sensitisation capability, in spite of the significant quenching experimentally observed (kq > 108 M−1s−1).134
Hence, the absolute value of TMP triplet energy has been estimated at ca. 310 kJ mol−1. According to the Sandros' equation, this is consistent with the observation of energy transfer from acetophenone (ET = 310 kJ mol−1), but not from 3-methoxyacetophenone (ET = 303 kJ mol−1).135
Benzophenone derivatives, with ET = 290 kJ mol−1, lower than that of TMP, have been shown to photosensitise Thd<>Thd formation at high nucleoside concentrations, in competition with a more favoured Paterno–Büchi photocycloaddition.31 This explains the efficient quenching (kq = 5.1 × 108 M−1s−1) observed in acetonitrile. Remarkably, LFP studies on (S)- and (R)-KP have shown a significant enantiodifferentiation in the quenching rate constants by Thd.55
In DNA, the photosensitiser triplet energy required for Pyr<>Pyr formation has been progressively shifted from ca. 300 kJ mol−1 (methoxyacetophenones)134 down to 290 kJ mol−1 (benzophenone and phthalimidine derivatives)31,48,55,68 and more recently to 267 kJ mol−1 (FQs).71,72 A series of fluoroquinolones (FQs), including ENX, pefloxacin (PFX), NFX, ANFX, OFX and rufloxacin (RFX) have been investigated to determine their potential as photosensitisers for Pyr<>Pyr dimers formation in DNA. At FQ concentrations for Pyr<>Pyr dimers formation in DNA. At FQ concentrations and light doses insufficient to produce direct single strand breaks, ENX, PFX and NFX are able to produce Pyr<>Pyr dimers in DNA as revealed by enzymatic treatment with T4 endonuclease V. By contrast, ANFX, OFX and RFX are inefficient in this assay (Fig. 9). This information has been combined with the absolute values of the triplet energies of ENX, PFX, NFX, ANFX, OFX and RFX, estimated by means of LFP, using flurbiprofen (FBP), 4-biphenylcarboxylic acid (BPC) and naproxen (NP) as energy acceptors (Table 6).71,72 All the results indicate that the threshold ET value required for a given compound to become a potential DNA photosensitiser via Thy<>Thy formation is in the range defined by the triplet energies of NFX and ANFX (265–269 kJ mol−1, see Fig. 9).
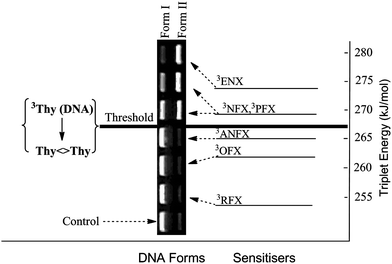 | ||
| Fig. 9 Triplet energy of photosensitisers and electrophoretic analysis of DNA Form I (supercoiled native form) and Form II (single strand break) obtained from mixtures containing pBR322 and FQs (20 μM) after 15 min of irradiation and subsequent T4 Endo V treatment. Adapted from Lhiaubet-Vallet et al.72 | ||
| 3ENX | 3PFX | 3NFX | 3ANFX | 3OFX | 3RFX | |
|---|---|---|---|---|---|---|
| a Quenching rate constants obtained in N2O-purged medium using 0.1 to 10 mM concentrations of the quenchers. b ΔE is the energy difference between the triplet excited states of quenchers and FQs obtained from Sandros' equation; kET = (kmax × e −ΔE/RT)/(e−ΔE/RT + 1) assuming that kmax = 2.2 × 109M−1 s−1. c Estimated values taking into account that ET of BPC and NP are ca. 266 and 259 kJ mol−1, respectively. | ||||||
| k ET (FBP)/109 (M−1 s−1)a | 0.3 | 0.09 | 0.09 | <0.01 | <0.01 | <0.01 |
| ΔEFBP-X (kJ mol−1)b | +4 | +8 | +8 | >+8 | >+8 | >+8 |
| k ET (BPC)/109 (M−1s−1)a | 2.3 | 1.7 | 1.5 | 0.9 | 0.02 | <0.01 |
| ΔEBPC-X (kJ mol−1)b | <−8 | −3 | −3 | +1 | >+8 | >+8 |
| k ET (NP)/109 (M−1s−1)a | 2.3 | 2.2 | 2.2 | 2.1 | 1.8 | 0.2 |
| ΔENP-X (kJ mol−1)b | <−8 | <−8 | <−8 | −7 | −4 | +6 |
| E T (kJ mol−1)c | 273 | 269 | 269 | 265 | 262 | 253 |
Moreover, when the Phs ET is lower than the threshold, triplet quenching by Pyr derivatives can only occur by pathways not involving energy transfer. This is the case for menadione, whose ET is ca. 243 kJ mol−1. The high quenching rate constants (between 1.0 and 2.5 × 109 M−1s−1) correspond to electron transfer, generating menadione radical anion and Pyr radical cations (Scheme 3).144
As stated above, in the case of Cyt, dCyd and dCMP, the triplet energy is higher than that reported for Thy, Thd and TMP. Therefore their triplet excited states are difficult to detect when both types of Pyr units are present, owing to deactivation via base-to-base energy transfer. Nonetheless, the transient absorption spectra have been recorded using acetone as Phs. From this type of experiment, it has been possible to determine the rate constants of unimolecular decay (k0), as well as those of self quenching (kS) (Table 7).133
6. Summary and outlook
Triplet excited states play a key role in the dimerisation of pyrimidine bases, not only in photosensitised processes, but also upon direct UV-irradiation. Cyclobutane pyrimidine dimers (CPDs) are by far the most relevant Pyr photoproducts obtained by sensitisation. Spore photoproducts may also be formed through this pathway; however, they are not found in mammals or other higher organisms. By contrast, there is no evidence for photosensitised reactions leading to pyrimidine (6–4) pyrimidone photoproducts. The mechanism of direct UVA-induced CPDs formation is still controversial; as DNA hardly absorbs in this wavelength range, the involvement of endogenous photosensitisers cannot be safely ruled out. In spite of the importance of the triplet pathway in Pyr photodimerisation, further efforts are needed to achieve a better understanding by means of theoretical calculations. Issues such as the lack of pyrimidine (6–4) pyrimidone formation or the special conditions required to obtain spore photoproducts from the triplet manifold should be explained. Finally, the triplet energy of thymine in DNA has been found to be much lower than that of the free base or the nucleoside. As this is a key parameter to anticipate the potential photocarcinogenicity of photosensitisers, it seems interesting to clarify how the various structural features (sequence, π-stacking, base pairing, etc.) modulate its actual value in the different microenvironments of the biomacromolecule.Acknowledgements
Financial support from the Spanish Government (CTQ2009-13699, CTQ2009-14196, JAE Doc fellowship for M.C.C. and Ramon y Cajal contract for V. L.-V.) and EU (CM0603) is gratefully acknowledged.Notes and references
- T. L. Oncology, Lancet Oncol., 2009, 10, 835 CrossRef.
- T. Who, Fact Sheet n° 287, 2010.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- R. D. Ley, Cancer Res., 1997, 57, 3682–3684 CAS.
- R. B. Setlow, E. Grist, K. Thompson and D. Woodhead, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6666–6670 CAS.
- P. J. Rochette, J.-P. Therrien, R. Drouin, D. Perdiz, N. Bastien, E. A. Drobetsky and E. Sage, Nucleic Acids Res., 2003, 31, 2786–2794 CrossRef CAS.
- D. L. Mitchell, A. A. Fernandez, R. S. Nairn, R. Garcia, L. Paniker, D. Trono, H. D. Thames and I. Gimenez-Conti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9329–9334 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- A. R. Young, C. S. Potten, O. Nikaido, P. G. Parsons, J. Boenders, J. M. Ramsden and C. A. Chadwick, J. Invest. Dermatol., 1998, 111, 936–940 CrossRef CAS.
- M. S. Cooke, M. D. Evans, R. M. Burd, K. Patel, A. Barnard, J. Lunec and P. E. Hutchinson, J. Invest. Dermatol., 2001, 116, 281–285 CrossRef CAS.
- S. Mouret, C. Phillippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, Org. Biomol. Chem., 2010, 8, 1706–1711 RSC.
- Y. Jiang, M. Rabbi, M. Kim, C. H. Ke, W. Lee, R. L. Clark, P. A. Mieczkowski and P. E. Marszalek, Biophys. J., 2009, 96, 1151–1158 CrossRef CAS.
- R. M. Tyrrell and S. M. Keyse, J. Photochem. Photobiol., B, 1990, 4, 349–361 CrossRef CAS.
- A. Besaratinia, T. W. Synold, H.-H. Chen, C. Chang, B. Xi, A. D. Riggs and G. P. Pfeifer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10058–10063 CrossRef CAS.
- Z. Kuluncsics, D. Perdiz, E. Brulay, B. Muel and E. Sage, J. Photochem. Photobiol., B, 1999, 49, 71–80 CrossRef CAS.
- D. Perdiz, P. Gróf, M. Mezzina, O. Nikaido, E. Moustacchi and E. Sage, J. Biol. Chem., 2000, 275, 26732–26742 CAS.
- J. Cadet, S. Courdavault, J. L. Ravanat and T. Douki, Pure Appl. Chem., 2005, 77, 947–961 CrossRef CAS.
- R. Costalat, J. Blais, J. P. Ballini, A. Moysan, J. Cadet, O. Chalvet and P. Vigny, Photochem. Photobiol., 1990, 51, 255–262 CrossRef CAS.
- A. Moysan, A. Viari, P. Vigny, L. Voituriez, J. Cadet, E. Moustacchi and E. Sage, Biochemistry, 1991, 30, 7080–7088 CrossRef CAS.
- R. S. Stern, L. E. J. and L. Väkevä, J. Natl. Cancer Inst., 1998, 90, 1278–1284 CrossRef CAS.
- A. R. Young, J. Photochem. Photobiol., B, 1990, 6, 237–247 CrossRef CAS.
- T. E. Spratt, S. S. Schultz, D. E. Levy, D. Chen, G. Shlueter and G. M. Williams, Chem. Res. Toxicol., 1999, 12, 809–815 CrossRef CAS.
- S. Sauvaigo, T. Douki, F. Odin, S. Caillat, J. L. Ravanat and J. Cadet, Photochem. Photobiol., 2001, 73, 230–237 CrossRef.
- M. C. Cuquerella, F. Bosca, M. A. Miranda, A. Belvedere, A. Catalfo and G. De Guidi, Chem. Res. Toxicol., 2003, 16, 562–570 CrossRef CAS.
- V. Lhiaubet-Vallet, F. Bosca and M. A. Miranda, Photochem. Photobiol., 2009, 85, 861–868 CrossRef CAS.
- M. Mäkinen, P. D. Forbes and F. Stenbäck, J. Photochem. Photobiol., B, 1997, 37, 182–187 CrossRef CAS.
- G. Klecak, F. Urbach and H. Urwyler, J. Photochem. Photobiol., B, 1997, 37, 174–181 CrossRef CAS.
- B. E. Jonhson, N. K. Gibbs and J. Ferguson, J. Photochem. Photobiol., B, 1997, 37, 171–173 CrossRef CAS.
- T. Itoh, H. Miyauchi-Hashimoto, A. Sugihara, K. Tanaka and T. Horio, J. Invest. Dermatol., 2005, 125, 554–559 CrossRef CAS.
- K. Sandros, Acta Chem. Scand., 1964, 18, 2355–2374.
- S. Encinas, N. Belmadoui, M. J. Climent, S. Gil and M. A. Miranda, Chem. Res. Toxicol., 2004, 17, 857–862 CrossRef CAS.
- H. Morrison and R. Kleopfer, J. Am. Chem. Soc., 1968, 90, 5037–5038 CrossRef CAS.
- P. J. Wagner and D. J. Bucheck, J. Am. Chem. Soc., 1970, 92, 181–185 CrossRef CAS.
- J. Cadet, L. Voituriez, F. E. Hruska, L.-S. Kan, F. A. A. de Leeuw and C. Altona, Can. J. Chem., 1985, 63, 2861–2868 CAS.
- A. J. Varghese, Photochem. Photobiol., 1972, 15, 113–118 CAS.
- A. A. Lamola, Photochem. Photobiol., 1968, 7, 619–632 CrossRef CAS.
- C. L. Greenstock and H. E. Johns, Biochem. Biophys. Res. Commun., 1968, 30, 21–27 CrossRef CAS.
- S. R. Aliwell, B. S. Martincigh and L. F. Salter, J. Photochem. Photobiol., A, 1993, 71, 137–146 CrossRef CAS.
- R. Kleopfer and H. Morrison, J. Am. Chem. Soc., 1972, 94, 255–264 CrossRef.
- N. Chouini-Lalanne, M. Defais and N. Paillous, Biochem. Pharmacol., 1998, 55, 441–446 CrossRef CAS.
- M. L. Meistrich and A. A. Lamola, J. Mol. Biol., 1972, 66, 83–95 CrossRef CAS.
- A. A. Lamola, M. Gueron, T. Yamane, J. Eisinger and R. G. Shulman, J. Chem. Phys., 1967, 47, 2210–2217 CrossRef CAS.
- P. I. Hønnas and H. B. Steen, Photochem. Photobiol., 1970, 11, 67–76 CrossRef CAS.
- I. von Wilucki, H. Matthäus and C. H. Krauch, Photochem. Photobiol., 1967, 6, 497–500 CrossRef.
- D. Elad, C. Krüger and G. M. J. Schmidt, Photochem. Photobiol., 1967, 6, 495–496 CrossRef CAS.
- B. H. Jennings, S.-C. Pastra and J. L. Wellington, Photochem. Photobiol., 1970, 11, 215–226 CrossRef CAS.
- E. Ben-Hur, D. Elad and R. Ben-Ishai, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1967, 149, 355–360 Search PubMed.
- T. Delatour, T. Douki, C. D'Ham and J. Cadet, J. Photochem. Photobiol., B, 1998, 44, 191–198 CrossRef CAS.
- T. Douki, M. Court and J. Cadet, J. Photochem. Photobiol., B, 2000, 54, 145–154 CrossRef CAS.
- N. Belmadoui, S. Encinas, M. J. Climent, S. Gil and M. A. Miranda, Chem.–Eur. J., 2006, 12, 553–561 CrossRef.
- G. Prakash and D. E. Falvey, J. Am. Chem. Soc., 1995, 117, 11375–11376 CrossRef CAS.
- K. Nakatani, T. Yoshida and I. Saito, J. Am. Chem. Soc., 2002, 124, 2118–2119 CrossRef CAS.
- A. J. Varghese, Photochem. Photobiol., 1975, 21, 147–151 CrossRef CAS.
- J. Trzcionka, V. Lhiaubet-Vallet, C. Paris, N. Belmadoui, M. J. Climent and M. A. Miranda, ChemBioChem, 2007, 8, 402–407 CrossRef CAS.
- V. Lhiaubet-Vallet, S. Encinas and M. A. Miranda, J. Am. Chem. Soc., 2005, 127, 12774–12775 CrossRef CAS.
- M. E. Umlas, W. A. Frankling, G. L. Chan and W. A. Haseltine, Photochem. Photobiol., 1985, 42, 265–273 CrossRef CAS.
- F.-T. Liu and N. C. Yang, Biochemistry, 1978, 17, 4865–4876 CrossRef CAS.
- W. Mu, Q. Han, Z. Luo and Y. Wang, Anal. Biochem., 2006, 353, 117–123 CrossRef CAS.
- M. Kaneko, A. Matsuyama and C. Nagata, Nucleic Acids Res., 1979, 6, 1177–1187 CrossRef CAS.
- M. W. Logue and N. J. Leonard, J. Am. Chem. Soc., 1972, 94, 2842–2846 CrossRef CAS.
- T. M. G. Koning, J. J. G. van Soest and R. Kaptein, Eur. J. Biochem., 1991, 195, 29–40 CrossRef CAS.
- N. J. Leonard, R. S. McCredie, M. W. Logue and R. L. Cundall, J. Am. Chem. Soc., 1973, 95, 2320–2324 CrossRef CAS.
- J. Yamamoto, K. Nishiguchi, K. Manabe, C. Masutani, F. Hanaoka and S. Iwai, Nucleic Acids Res., 2010, 1–11.
- R. O. Rahn and L. C. Landry, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1971, 247, 197–206 Search PubMed.
- J. L. Hosszu and R. O. Rahn, Biochem. Biophys. Res. Commun., 1967, 29, 327–330 CrossRef CAS.
- R. B. Setlow and W. L. Carrier, J. Mol. Biol., 1966, 17, 237–254 CrossRef CAS.
- V. Lhiaubet, N. Paillous and N. Chouini-Lalanne, Photochem. Photobiol., 2001, 74, 670–678 CAS.
- V. Lhiaubet-Vallet, J. Trzcionka, S. Encinas, M. A. Miranda and N. Chouini-Lalanne, J. Phys. Chem. B, 2004, 108, 14148–14153 CrossRef CAS.
- F. Bourre, G. Renault, P. C. Seawell and A. Sarasin, Biochimie, 67, pp. 293–299 Search PubMed.
- J. Trzcionka, V. Lhiaubet-Vallet and N. Chouini-Lalanne, Photochem. Photobiol. Sci., 2004, 3, 226–230 RSC.
- F. Bosca, V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell and M. A. Miranda, J. Am. Chem. Soc., 2006, 128, 6318–6319 CrossRef CAS.
- V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell, F. Bosca and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 7409–7414 CrossRef CAS.
- L. Marrot, J. P. Belaidi, C. Jones, P. Perez, L. Riou, S. A. and J. R. Meunier, J. Invest. Dermatol., 2003, 121, 596–606 CrossRef CAS.
- N. J. Traynor and N. K. Gibbs, Photochem. Photobiol., 1999, 70, 957–959 CAS.
- A. A. Lamola, Pure Appl. Chem., 1970, 24, 599–610 CrossRef CAS.
- A. A. Lamola and T. Yamane, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 443–446 CrossRef CAS.
- M. H. Patrick and J. M. Snow, Photochem. Photobiol., 1977, 25, 373–384 CrossRef CAS.
- L. A. Guillo, J. Blais, P. Vigny and A. Spassky, Photochem. Photobiol., 1995, 61, 331–335 CrossRef CAS.
- K. S. Robinson, N. J. Traynor, H. Moseley, J. Ferguson and J. A. Woods, Toxicol. in Vitro, 2010, 24, 1126–1132 CrossRef CAS.
- N. J. Traynor, B. Kratzer, M. A. Miranda and N. K. Gibbs, Br. J. Dermatol., 1999, 140, 784.
- L. Marrot and J.-R. Meunier, J. Am. Acad. Dermatol., 2008, 58, S139–S148 CrossRef.
- P. Walrant, R. Santus and M. Charlier, Photochem. Photobiol., 1976, 24, 13–19 CrossRef CAS.
- K. Bolton, B. S. Martincigh and L. F. Salter, J. Photochem. Photobiol., A, 1992, 63, 241–248 CrossRef CAS.
- S. R. Aliwell, B. S. Martincigh and L. F. Salter, J. Photochem. Photobiol., A, 1993, 71, 147–153 CrossRef CAS.
- C. Desnous, D. Guillaume and P. Clivio, Chem. Rev., 2010, 110, 1213–1232 CrossRef CAS.
- J. E. J. Donnellan and R. B. Setlow, Science, 1965, 149, 308–310 CrossRef CAS.
- C. Mantel, A. Chandor, D. Gasparutto, T. Douki, M. Atta, M. Fontecave, P.-A. Bayle, J.-M. Mouesca and M. Bardet, J. Am. Chem. Soc., 2008, 130, 16978–16984 CrossRef CAS.
- T. Douki, B. Setlow and P. Setlow, Photochem. Photobiol. Sci., 2005, 4, 591–597 RSC.
- T. Douki, G. Laporte and J. Cadet, Nucleic Acids Res., 2003, 31, 3134–3142 CrossRef CAS.
- W. L. Nicholson, B. Setlow and P. Setlow, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8288–8292 CAS.
- B. Setlow and P. Setlow, Appl. Environ. Microbiol., 1993, 59, 640–643 CAS.
- R. O. Rahn and J. L. Hosszu, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1969, 190, 126–131 Search PubMed.
- T. Douki and J. Cadet, Photochem. Photobiol. Sci., 2003, 2, 433–436 RSC.
- A. J. Varghese, Biochemistry, 1970, 9, 4781–4787 CrossRef CAS.
- M. Gromova, E. Balanzat, B. Gervais, R. Nardin and J. Cadet, Int. J. Radiat. Biol., 1998, 74, 81–97 CrossRef CAS.
- A. A. Shaw and J. Cadet, J. Chem. Soc., Perkin Trans. 2, 1990, 2063–2070 RSC.
- M. T. Nguyen, R. Zhang, P.-C. Nam and A. J. Ceulemans, J. Phys. Chem. A, 2004, 108, 6554–6561 CrossRef CAS.
- M. Etinski, T. Fleig and C. M. Marian, J. Phys. Chem. A, 2009, 113, 11809–11816 CrossRef CAS.
- J. J. Serrano-Pérez, R. González-Luque, M. Merchán and L. Serrano-Andrés, J. Phys. Chem. B, 2007, 111, 11880–11883 CrossRef CAS.
- M. Etinski and C. M. Marian, Phys. Chem. Chem. Phys., 2010, 12, 4915–4923 RSC.
- M. Merchán, L. Serrano-Andrés, M. A. Robb and L. Blancafort, J. Am. Chem. Soc., 2005, 127, 1820–1825 CrossRef CAS.
- R. B. Zhang and L. A. Eriksson, J. Phys. Chem. B, 2006, 110, 7556–7562 CrossRef CAS.
- T. Climent, I. González-Ramírez, R. Gonzalez-Luque, M. Merchán and L. Serrano-Andres, J. Phys. Chem. Lett., 2010, 1, 2072–2076 Search PubMed.
- D. Roca-Sanjuán, G. Olaso-González, I. González-Ramírez, L. Serrano-Andrés and M. Merchán, J. Am. Chem. Soc., 2008, 130, 10768–10779 CrossRef CAS.
- R. Abouaf, J. Pommier, H. Dunet, P. Quan, P. C. Nam and M. T. Nguyen, J. Chem. Phys., 2004, 121, 11668–11674 CrossRef CAS.
- R. González-Luque, T. Climent, I. González-Ramírez, M. Merchán and L. Serrano-Andrés, J. Chem. Theory Comput., 2010, 6, 2103–2114 CrossRef CAS.
- T. Fleig, S. Knecht and C. Hättig, J. Phys. Chem. A, 2007, 111, 5482–5491 CrossRef CAS.
- T. Climent, R. González-Luque, M. Merchán and L. Serrano-Andrés, Chem. Phys. Lett., 2007, 441, 327–331 CrossRef CAS.
- A. M. Rasmussen, M. C. Lind, S. Kim and H. F. Schaefer, J. Chem. Theory Comput., 2010, 6, 930–939 CrossRef CAS.
- M. Boggio-Pasqua, G. Groenhof, L. V. Shäfer, H. Grubmüller and M. A. Robb, J. Am. Chem. Soc., 2007, 129, 10996–10997 CrossRef CAS.
- M. Merchán, R. González-Luque, T. Climent, L. Serrano-Andrés, E. Rodríguez, M. Reguero and D. Peláez, J. Phys. Chem. B, 2006, 110, 26471–26476 CrossRef CAS.
- B. Durbeej and L. A. Eriksson, J. Photochem. Photobiol., A, 2002, 152, 95–101 CrossRef CAS.
- J. J. Serrano-Perez, I. Gonzalez-Ramirez, P. B. Coto, M. Merchan and L. Serrano-Andres, J. Phys. Chem. B, 2008, 112, 14096–14098 CrossRef CAS.
- L. Blancafort and A. Migani, J. Am. Chem. Soc., 2007, 129, 14540–14541 CrossRef CAS.
- R. O. Rahn, R. G. Shulman and J. W. Longworth, Proc. Natl. Acad. Sci. U. S. A., 1965, 53, 893–896 CrossRef CAS.
- R. Bersohn and I. Isenberg, Biochem. Biophys. Res. Commun., 1963, 12, 205–208 CrossRef.
- R. Bersohn and I. Isenberg, J. Chem. Phys., 1964, 40, 3175–3180 CrossRef CAS.
- C. Hélène, Biochem. Biophys. Res. Commun., 1966, 22, 237–242 CrossRef CAS.
- R. O. Rahn, R. G. Shulman and J. W. Longworth, J. Chem. Phys., 1966, 45, 2955–2965 CrossRef CAS.
- J. Eisinger and R. G. Shulman, Science, 1968, 161, 1311–1319 CrossRef CAS.
- T. Montenay-Garestier and C. Hélène, Biochemistry, 1970, 9, 2865–2870 CrossRef CAS.
- H. Görner, J. Photochem. Photobiol., B, 1990, 5, 359–377 CrossRef CAS.
- V. Kleinwachter, J. Drobnik and L. Augenstein, Photochem. Photobiol., 1968, 7, 485–497 CrossRef CAS.
- J. J. Aaron, W. J. Spann and J. D. Winefordner, Talanta, 1973, 20, 855–865 CrossRef CAS.
- M. Guéron, J. Eisinger and R. G. Shulman, J. Chem. Phys., 1967, 47, 4077–4091 CrossRef CAS.
- I. Isenberg, R. Rosenbluth and S. L. J. Baird, Biophys. J., 1967, 7, 365–373 CrossRef CAS.
- C. Hélène and T. Montenay-Garestier, Chem. Phys. Lett., 1968, 2, 25–28 CrossRef CAS.
- J. Eisinger and R. G. Shulman, J. Mol. Biol., 1967, 28, 445–449 CrossRef CAS.
- R. S. Becker and G. Kogan, Photochem. Photobiol., 1980, 31, 5–13 CrossRef CAS.
- C. Salet, R. Bensasson and R. S. Becker, Photochem. Photobiol., 1979, 30, 325–329 CAS.
- C. Salet and R. Bensasson, Photochem. Photobiol., 1975, 22, 231–235 CrossRef CAS.
- K. Kasama, A. Takematsu and S. Arai, J. Phys. Chem., 1982, 86, 2420–2427 CrossRef CAS.
- Z.-h. Zuo, S.-d. Yao, J. Luo, W.-f. Wang, J.-s. Zhang and N.-y. Lin, J. Photochem. Photobiol., B, 1992, 15, 215–222 CrossRef CAS.
- I. G. Gut, P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 2366–2373 CrossRef CAS.
- P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 4256 CrossRef CAS.
- Q. H. Song, W. Z. Lin, S. D. Yao and N. Y. Lin, J. Photochem. Photobiol., A, 1998, 114, 181–184 CrossRef CAS.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Chem. Rev., 2004, 104, 1977–2019 CrossRef CAS.
- E. Samoylova, H. Lippert, S. Ullrich, I. V. Hertel, W. Radloff and T. Schultz, J. Am. Chem. Soc., 2005, 127, 1782–1786 CrossRef CAS.
- W. J. Schreier, T. E. Schrader, F. O. Koller, P. Gilch, C. E. Crespo-Hernández, V. N. Swaminathan, T. Carell, W. Zinth and B. Kohler, Science, 2007, 315, 625–629 CrossRef CAS.
- P. M. Hare, C. T. Middleton, K. I. Mertel, J. M. Herbert and B. Kohler, Chem. Phys., 2008, 347, 383–392 CrossRef.
- W. M. Kwok, C. Ma and D. L. Phillips, J. Am. Chem. Soc., 2008, 130, 5131–5139 CrossRef CAS.
- S. Marguet and D. Markovitsi, J. Am. Chem. Soc., 2005, 127, 5780–5781 CrossRef CAS.
- R. V. Bensasson, E. J. Land and T. G. Truscott, Laser Photolysis and Pulse Radiolysis, Oxford, 1983 Search PubMed.
- J. R. Wagner, J. E. van Lier and L. J. Johnston, Photochem. Photobiol., 1990, 52, 333–343 CAS.
| This journal is © The Royal Society of Chemistry 2011 |