A chemically-controlled supramolecular protein polymer formed by a myoglobin-based self-assembly system†
Koji
Oohora
a,
Akira
Onoda
ab,
Hiroaki
Kitagishi‡
a,
Hiroyasu
Yamaguchi
c,
Akira
Harada
c and
Takashi
Hayashi
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, 565-0871, Japan. E-mail: thayashi@chem.eng.osaka-u.ac.jp; Fax: + 81 6-6879-7930; Tel: + 81 6-6879-7928
bFrontier Research Base for Global Young Researchers, Graduate School of Engineering, Osaka University, Suita, 565-0871, Japan
cDepartment of Macromolecular Science, Graduate School of Science, Osaka University, Toyonaka, 560-0043, Japan
First published on 29th March 2011
Abstract
Artificial self-assembling systems comprised of proteins have the potential not only for mimicking naturally occurring protein clusters but also for creating functionalized supramolecular polymers. Here we report a new type of a supramolecular protein polymer which utilizes the original character and reactivity of the monomer protein. Myoglobin, an oxygen storage hemoprotein, was chosen as the monomer unit and was provided with an externally-attached heme on the protein surface which drives the formation of the fibrous supramolecular assembly through successive interprotein interactions between the external heme and the protein matrix. This assembly governed by myoglobin characteristics shows chemically-responsive stability and can be converted into extremely large protein clustersvia cross-linking. Interestingly, the assembly retains the oxygen storage function. Our present system can be used for construction of smart nanobiomaterials using various hemoproteins.
Introduction
Biomolecule-based synthetic nanostructures constructed by self-assembly are in high demand for development of biomaterials because of their biocompatibility, biodegradability, and defined morphology.1–9 Especially, protein assemblies have potential to be used in practical applications in fields such as tissue engineering, biomineralization, light harvesting, cascade reactions and drug delivery systems. Proteins may also prove to be attractive building blocks for artificial superstructures because their well-defined structures have high recognition ability and specific reactivity towards target molecules.4,10–13Artificial protein-based supramolecular nanostructures have been constructed by taking advantage of specific and/or non-specific interactions via combinations of chemical and biological techniques.12–21 Although much effort has been devoted to the development of “bottom-up” nanobiotechnology, most examples have involved simple assemblies with limited properties and functions. The new generation of nanobiomaterials will require reactivity, responsiveness to stimuli and the ability to expand into larger structures. A promising strategy for the development of such “smart biomaterials” is to integrate a protein function into the polymerized materials. Therefore, if a hemoprotein could be assembled with a highly ordered structure, the resulting cluster would be very attractive to use in the development of nanobiomaterials because the heme22prosthetic group has many diverse functions in biological processes such as electron transfer, catalysis and sensing. According to this concept, we have recently described a supramolecular hemoprotein polymer formed by multiple interprotein heme–heme pocket interactions using modified cytochrome b562, one of the simplest hemoproteins, as a monomer unit. This monomer was modified by removing the native heme and covalently linking an external heme to the surface of the resulting apo-cytochrome b562.23,24 Our next objective is to extend the supramolecular self-assembly system of cytochrome b562 to that of other hemoproteins with attractive physicochemical properties and reactivities.
To construct a new supramolecular protein nanostructure with specific properties and functions, we selected sperm whale myoglobin (Mb), an O2storage protein, as a building block. Our underlying strategy for construction of the supramolecular assembly is illustrated schematically in Fig. 1. One of the most important points to consider is whether the Mb-based supramolecular assembly (poly-1-Mb) retains sufficient affinity for O2. In addition, it is instructive to evaluate the relationships between various small molecules which are known to bind to the heme iron of Mb as a sixth ligand and to assess the thermodynamic stability of poly-1-Mb. Examples of such sixth ligands include H2O, CN−, O2 and CO. It is known that ligation of a sixth ligand can regulate the affinity of heme with respect to its interaction with the heme pocket.25
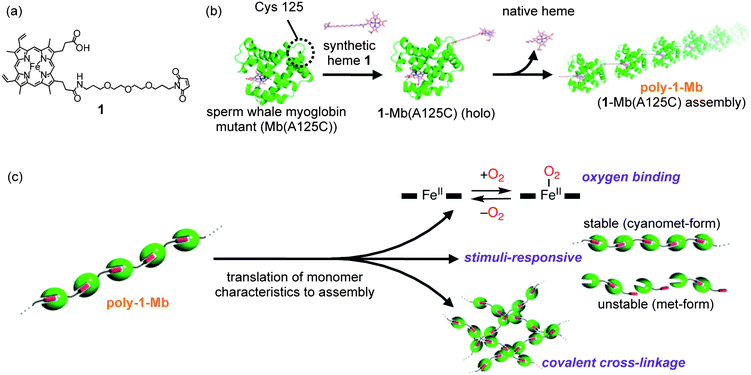 | ||
| Fig. 1 Schematic representation of supramolecular polymerization of myoglobin. (a) Structure of synthetic heme 1. This compound has regioisomers with respect to the substitution position in the two heme propionate side chains of protoheme IX. (b) Formation of myoglobin-based supramolecular assembly (poly-1-Mb) driven by the heme–heme pocket interaction. (c) Functionalization by translation of myoglobin characteristics to the supramolecular assembly. | ||
In this paper, we describe the preparation, characterization and chemical properties of a new supramolecular myoglobin polymer. Furthermore, in expanding this hemoprotein assembly to a higher dimensional structure, we demonstrate the construction of a large water-soluble myoglobin cluster where cross-linking of linear fibers of myoglobin is initiated upon addition of H2O2.26 Our goal is to rationally design a supramolecular hemoprotein polymer as a protein assembly with distinctive properties in order to contribute to the development of practical nanobiomaterials.
Results and discussion
Preparation and characterization of the myoglobin polymer
The single mutant of sperm whale myoglobin (Mb(A125C)), which has only one cysteine residue at the 125 position, is used in this investigation as a scaffold for the protein-based supramolecular assembly. As shown in Fig. 1a, we designed the synthetic heme 1 whose propionate side chain was modified by maleimide as a thiol-acceptor27 through the relatively short linker toward the linear protein polymerization preferentially without any cyclic oligomers. The covalent linkage of Mb(A125C) to synthetic heme 1 followed by removal of the native heme yields the apoprotein with an externally-attached heme (1-Mb(A125C)).28 The modified protein, 1-Mb(A125C), was characterized by deconvoluted electrospray ionization mass spectrometry (ESI-MS, Fig. S2, ESI†): 18259.7 for 1-Mb(A125C) (calcd. = 18260.9), whereas no peaks were observed for the unreacted Mb(A125C) and 1. In addition, Ellman's method29 (known as the reactive thiol assay), shows that the modified protein lacks the reactive thiol. This indicates that the thiol group of Cys125 reacts completely with 1 to yield 1-Mb(A125C).30 Furthermore, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using SDS buffer without 2-mercaptoethanol indicates that the disulfide dimer is absent after the surface modification reaction (Fig. S3, ESI†). Next, the existence of the heme–heme pocket interaction in this system was preliminarily supported by UV-vis spectral data (Fig. S4, ESI†). The UV-vis spectrum of a solution of 1-Mb(A125C) at neutral pH is essentially identical to that of the met-form (FeIII–H2O) of native Mb (nMb). Furthermore, the oxy-form of 1-Mb(A125C) was also prepared using the usual procedure for preparation of oxy-nMb31 and the UV-vis spectrum of oxy-form of 1-Mb(A125C) was found to have the characteristic Soret band (417 nm) and Q bands (543 and 580 nm), which indicate the oxy-form (FeII–O2) of nMb. Although the perturbation of the protein structure often accelerates the autoxidation of the oxygenated species,31 the oxy-form of 1-Mb(A125C) remains stable under ambient conditions (vide infra). This observation provides strong support for our interpretation that the externally-attached heme on the protein surface is successfully incorporated into the heme pocket of another Mb(A125C) monomer to produce the interprotein assembly as intended.To further confirm the existence of the protein assembly, the solution of 1-Mb(A125C) was analyzed by size exclusion chromatography (SEC).32Fig. 2a shows the SEC profiles of the Mb(A125C) monomer, the Mb(A125C) disulfide dimer and 1-Mb(A125C). The Mb(A125C) monomer and the disulfide dimer are eluted from the column with volumes of 17.0 and 15.5 mL respectively, whereas 1-Mb(A125C) elutes much earlier. Dissociation of this assembly by acid denaturation (pH 4.8) or addition of one equivalent of native hemin showed the peak of the completely dissociated protein at 17.0 mL (Fig. S5, ESI†). These results indicate that a large stable supramolecular myoglobin assembly, herein designated poly-1-Mb, has been formed viaheme–heme pocket interactions. The observed broad SEC profile is a typical observation of equilibrium polymerization systems such as the previously reported cytochrome b562 assembly.23 The molecular size distribution of the poly-1-Mb is clearly dependent on the concentration of 1-Mb(A125C) in the solution (Fig. 2b)(Table S1, ESI†). Dilution of poly-1-Mb delays its elution in the SEC profile with a concomitant increase of the low molecular weight components. Therefore, this finding supports our proposal that the concentration-dependent polymerization is controlled by thermodynamic equilibrium of the interprotein heme–heme pocket interactions.
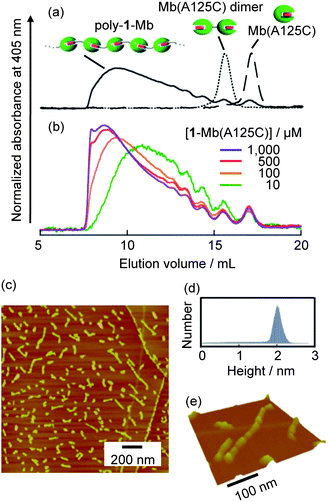 | ||
| Fig. 2 Characterization of poly-1-Mb. (a) SEC traces of poly-1-Mb, Mb(A125C) dimer and Mb(A125C). (b) SEC traces of poly-1-Mb at various concentrations of 1-Mb(A125C). (c) AFM image of poly-1-Mb on the HOPG substrate. (d) Height distribution of the image in (c). (e) 3D representation of an enlarged image of (c). | ||
The structure of poly-1-Mb deposited on a highly oriented pyrolytic graphite (HOPG) substrate has been directly imaged by atomic force microscopy (AFM).33 As shown in Fig. 2c, many linear assemblies with various lengths were observed. The monodiverse height diagram (ca. 2 nm, Fig. 2d) indicates the presence of a single protein chain of poly-1-Mb. The heights of the objects are slightly smaller than the heights within the range of 2.5–5.0 nm which are expected on the basis of the crystal structure of nMb.34 This is most likely due to interactions of the specific surface region of poly-1-Mb with the hydrophobic HOPG substrate or the occurrence of slight structural changes during dry-up. In a closer view of the selected linear objects (Fig. 2e), it can be seen that the fiber reaches a length of ca. 450 nm. This corresponds to the length of an 80-mer protein.
Stability of the supramolecular polymer controlled by axial ligation
Various axial sixth ligands and oxidation states of heme affect heme binding properties for the protein matrix of Mb.25 Therefore, we expected that the thermodynamic assembly behavior of poly-1-Mb would be influenced by the state of axial ligation of the heme iron and the electronic structure of the heme moiety of each Mb unit. Fig. 3 shows the results of fractionation experiments performed to investigate the stability of the supramolecular assemblies.12,23 A concentrated poly-1-Mb solution (0.6 mM per heme moiety) was injected onto the SEC column and an early-eluted fraction (indicated by the shaded region in the top panel of Fig. 3) was collected. Immediate reanalysis of the separated fraction revealed that the polymeric population, for the most part, retained its original size. However, after standing at 25 °C for 24 h, the SEC profile was observed to return to the initial broad distribution because of the thermodynamic equilibrium of poly-1-Mb. In contrast, when two equivalents of potassium cyanide were added to the fractionated poly-1-Mb solution, the polymeric population was found to be retained after standing at 25 °C for 24 h (Fig. 3d). This clearly indicates that the cyanomet-form (FeIII–CN−) of poly-1-Mb is stabilized by coordination of CN− to the heme iron. The result can be rationalized by the known fact that the strength of the interactions between the externally attached heme and the heme pocket in poly-1-Mb increases as a result of π–π* back-donation from the heme iron to the CN− ligand. This in turn leads to strengthening of the His93–Fe bond.25 Similarly, additional experiments show that the oxidation state of the heme iron can also modulate the stability of poly-1-Mb. The deoxy-form of poly-1-Mb was prepared immediately after fractionation of the polymeric species by reduction of poly-1-Mb using sodium dithionite in a glove box under a N2 atmosphere (O2 < 1 ppm). This deoxy species of poly-1-Mb, for the most part, retains its original size. In addition, the CO-form prepared by subjecting the deoxy-form of poly-1-Mb to 1 atm of CO has stability similar to that of the deoxy- and cyanomet-forms. These results are consistent with the expected stabilization of the His93–Fe bond in the ferrous state of nMb.25 These findings allow us to conclude that the stability of the supramolecular hemoprotein polymer can be conveniently regulated by addition of exogenous ligands which bind to the axial sixth position of the heme within the heme pocket.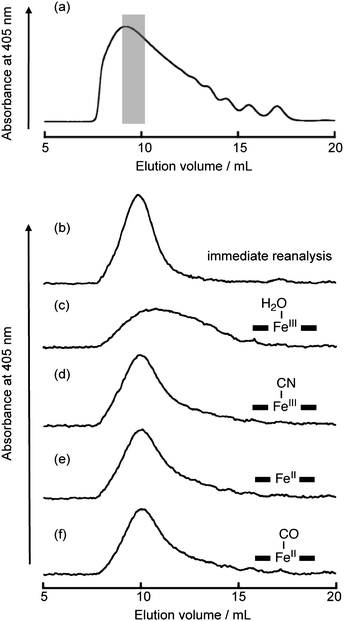 | ||
| Fig. 3 SEC traces of poly-1-Mb for the fractionation experiments to estimate stability changes by heme ligand and oxidation state. (a) SEC trace of met-form of poly-1-Mb. The fraction of the shaded area was collected. (b) Immediately reanalyzed trace of met-form. Reanalyzed traces after standing at 25 °C for 24 h: (c) met-, (d) cyanomet-, (e) deoxy- and (f) CO-forms of poly-1-Mb, respectively. Each form was prepared soon after separation and schematically represented by the corresponding inset. | ||
Cross-linking of the myoglobin polymers
It is known that a covalently linked Mb dimer can be formed upon addition of H2O2 to a solution of nMbvia a radical coupling reaction, which takes place on the protein surface due to the generation of the free radicals derived from the reaction of H2O2 with the heme iron.26 Therefore, we have exploited this knowledge for development of a cross-linked version of poly-1-Mb because a fibrous protein polymer has a potential to be used in the development of practical biomaterials by cross-linking of its structure.35 As a result, the reaction of poly-1-Mb with 1.5 equivalents of H2O2 produced high molecular weight components. Surprisingly, it was found that essentially all of the assemblies reached the exclusion limit (ca. 1300 kDa) of the Superdex 200 column (Fig. 4). Furthermore, the SDS-PAGE exhibits bands, which indicate the existence of covalently dimerized 1-Mb(A125C) (36 kDa) after the reaction (Fig. S6, ESI†). These results indicate that the protein radicals, which are formed by self-peroxidation, are coupled to yield a cross-linked form of poly-1-Mb, which completely dissociated to a monomer and covalently linked dimer under denaturing conditions. We propose that the formation of the cross-linked species occurs at tyrosine residues in a manner similar to the cross-linking of nMb (Tyr153 with Tyr153′ or Tyr140′ of another molecule of nMb) according to a previous report.26 This site-specific cross-linking reaction on the Mb surface is clearly distinguished from the usual one by glutaraldehyde as a cross-linker, which brings about oligomerization of proteins through ramdom coupling with some reactive primary amines such as Lys residues and terminal amine. In contrast, no oligomeric protein is obtained upon the addition of H2O2 into a solution of nMb,26 suggesting that it is necessary to have the fibrous protein first as a precursor to obtain the three-dimensional Mb cluster (vide infra). Support for the formation of cross-linked poly-1-Mb is provided directly from the observation of characteristic images by field emission scanning electron microscopy (SEM). The SEM images indicate the presence of detailed structures on the HOPG substrate (Fig. 5). Sub-micrometre-sized network structures were observed and the widths of the fibers in the networks ranged from ca. 4–9 nm. This range is consistent with the sizes of one to two molecules of nMb as estimated from the crystal structure of nMb (Fig. 5c). Therefore, these network-shaped objects provide proof of the formation of cross-linked polymers by the reaction of poly-1-Mb with H2O2. To the best of our knowledge, the present system provides the first report of cross-linkage of supramolecular protein fibers by self-activation, that is, generation of protein radicals via the reaction of the heme iron with H2O2 in the Mb matrix.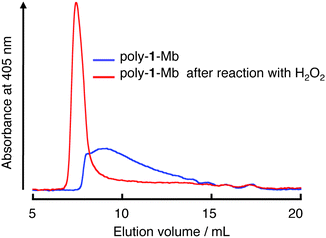 | ||
| Fig. 4 SEC traces of poly-1-Mb and cross-linked poly-1-Mb. | ||
 | ||
| Fig. 5 Microscopic characterization of cross-linked poly-1-Mb. (a) SEM image of cross-linked poly-1-Mb. (b) Enlarged SEM images. Measured fiber widths are shown in (b). (c) Plausible structural details of the network-shaped objects. | ||
O2 affinities of myoglobin polymers
In general, protein modification would be expected to cause a loss of the intrinsic protein functions due to even slight conformational changes.31 Therefore, we evaluated the kinetic parameters related to O2 binding for deoxygenated poly-1-Mb and cross-linked poly-1-Mb using conventional laser flash photolysis and stopped-flow rapid mixing techniques. The O2 affinities determined for poly-1-Mb and its cross-linked structure were 5.0 × 105 M−1 and 4.8 × 105 M−1, respectively. These values are similar to the O2 affinity for nMb (8.6 × 105 M−1). In particular, the O2 dissociation rate constants for poly-1-Mb and cross-linked poly-1-Mb are consistent with the O2 dissociation rate constant observed for nMb. This indicates that the oxygenated species are fully stabilized in the assembled Mb (Table S2, ESI†). In contrast, the slight decrease in the rate of O2 association for Mb assemblies may be caused by the amidation of the heme-propionate side chain, because it is known that an amide-modified heme-propionate side-chain generally reduces the access of O2 into the protein matrix36 (Fig. S7 and Table S2, ESI†). These findings support our conclusion that the polymerization of myoglobin does not have a significant influence on the protein structure. This is surprising considering that even small structural deviations can cause drastic changes in ligand binding parameters.Conclusions
The present results demonstrate a new strategy for supramolecular hemoprotein polymerization in which an interprotein heme–heme pocket interaction can be developed using apomyoglobin with an external covalently-linked heme moiety. The self-assembly behavior indicates that the polymerization of myoglobin is controlled by the affinity of the externally-linked heme for the heme pocket of another myoglobin monomer. Moreover, we demonstrate the first example of construction of a large three-dimensional protein network derived from cross-linkage of fibrous myoglobin polymers initiated by protein self-activation using H2O2. It is further noted that the physiological function of myoglobin is substantially retained in the polymeric species. This work may serve as an illustration of an attractive strategy for design and construction of supramolecular nanobiomaterials such as catalysts, sensors, and small molecule storage units based upon hemoprotein monomers.Experimental procedures
Preparation of poly-1-Mb
The modified heme 1 (2.0 μmol, 10 eq.) in dimethyl sulfoxide/H2O solution (800 μL, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) including 100 mM L-histidine was added to 100 μM of Mb(A125C) monomer in 100 mM aqueous L-histidine solution (2 mL). The mixture was then gently stirred at room temperature, under an atmosphere of N2 in a glove box (O2 < 1 ppm). The reaction mixture was then dialyzed against a 100-fold volume of 10 mM phosphate buffer, pH 6.0, at 4 °C for 1 h. After the dialysis, the solution was acidified to pH 2.0 with an aqueous HCl solution. The free heme (1 and the native heme) was removed from the sample solutions by extraction with 2-butanone (5 mL × 4).28 The separated aqueous layer was dialyzed against a 100-fold volume of 100 mM phosphate buffer, pH 7.0, at 4 °C (2 h × 4). The resulting protein solution was concentrated by ultrafiltration to a heme concentration of 10−3 M, and the solution was stored in the dark at 4 °C.
:1, v/v) including 100 mM L-histidine was added to 100 μM of Mb(A125C) monomer in 100 mM aqueous L-histidine solution (2 mL). The mixture was then gently stirred at room temperature, under an atmosphere of N2 in a glove box (O2 < 1 ppm). The reaction mixture was then dialyzed against a 100-fold volume of 10 mM phosphate buffer, pH 6.0, at 4 °C for 1 h. After the dialysis, the solution was acidified to pH 2.0 with an aqueous HCl solution. The free heme (1 and the native heme) was removed from the sample solutions by extraction with 2-butanone (5 mL × 4).28 The separated aqueous layer was dialyzed against a 100-fold volume of 100 mM phosphate buffer, pH 7.0, at 4 °C (2 h × 4). The resulting protein solution was concentrated by ultrafiltration to a heme concentration of 10−3 M, and the solution was stored in the dark at 4 °C.
Reaction of poly-1-Mb with H2O2
The cross-linking reaction was carried out in 100 mM potassium phosphate buffer at pH 7.0. The solution of H2O2 (2.3 mM, 4 μL, 1.5 eq with respect to heme 1) was added to the poly-1-Mb stock solution (0.3 mM, 20 μL). After a 2 h incubation period at 25 °C, the reaction mixture was allowed to stand at 4 °C for 1 day.SEC analyses
For SEC analyses, 100 mM potassium phosphate buffer, pH 7.0, was used as the eluent. The analyses were performed at 4 °C at a flow rate of 0.5 mL min−1 with monitoring of the absorbance at 405 nm for detection. The Superdex 200 column was calibrated using the following reagents: Blue Dextran (2000 kDa, used to determine the void volume of the column), ferritin (400 kDa), catalase (232 kDa), albumin (67 kDa), and chymotrypsinogen (25 kDa).AFM measurements
The samples were prepared as follows: a freshly cleaved HOPG substrate was immersed in the poly-1-Mb solution (ca. 10−8 M, in 100 mM phosphate buffer, pH 7.0), which was prepared just prior to the immersion of the concentrated poly-1-Mb solution (ca. 10−3 M, in 100 mM phosphate buffer, pH 7.0) to minimize the dissociation from the polymer into monomers by equilibration in the diluted state. After immersion for a few seconds, the sample plate was picked up and the surface was extensively rinsed with H2O. The HOPG substrate was allowed to stand for a few hours in a desiccator with CaCl2 for drying the sample, before it was transferred onto the stage of the AFM. For the measurements, we used the tapping mode AFM with a SiN4 tip having a radius of ca. 10 nm.37 The line scan speed was 2 Hz with 512 pixels per line.SEM measurements
To a freshly cleaved HOPG substrate was dropped 10 μL of the cross-linked poly-1-Mb solution (ca. 10−9 M, in deionized water), which was desalted from the sample solution (100 mM phosphate buffer, pH 7.0) using a Hi-Trap™ desalting column (GE-Healthcare) just prior to the drop to minimize the nonspecific aggregation under low ionic strength conditions. When the dropping was complete, the sample plate was immediately immersed in liquid nitrogen and picked up to lyophilize the protein solution in vacuo. Images were taken using an ultra resolution cold field emission SEM under conditions of low acceleration voltage (1.5 kV) because the samples were not stained.O2 binding of Mbs
To prepare oxygenated Mbs (oxy-nMb, oxy-poly-1-Mb or oxy-cross-linked poly-1-Mb), met-Mbs were reduced by sodium dithionite to yield deoxy-Mbs, which were then purified from excess sodium dithionite using a Sephadex G-25 gel filtration column with 100 mM phosphate buffer, pH 7.0. During the elution, deoxy-Mb species were smoothly converted to the oxy-Mbs by O2 dissolved in the buffer. The association of O2 was observed by the absorbance changes at 435 nm after the flash photolysis of the oxy form under 1 atm of air at 25 °C. The time course of the absorbance was analyzed by single phase kinetics to give a pseudo-first-order rate constant. By dividing this value by the O2 concentration (2.64 × 10−4 M), we obtained the association rate constant. The dissociation of O2 was assessed by following the UV-vis spectral changes after rapidly mixing the oxygenated form with excess K3[Fe(CN)6] using a stopped flow apparatus. The O2 affinity was calculated from the ratio of the association and dissociation rates.Autoxidation for oxy-Mbs
Autoxidation reactions of oxy-Mbs to the corresponding met-forms were monitored by absorbance changes in the range of 500–700 nm every 30 min at 37 °C under aerobic conditions. The time course of the absorbance at 580 nm was analyzed by the first-order kinetics to indicate the autoxidation rate.Acknowledgements
We express our gratitude to Professor Shun Hirota (Nara Institute of Science and Technology) for providing the A125C myoglobin mutant expression system. We gratefully acknowledge Dr. M. Konomi, Dr. S. Watanabe and Mr. Y. Orai (Hitachi High-Technology Co.) for assistance with SEM measurements and helpful discussions. We are grateful for financial supports from the Asahi Glass Foundation, Japan, the Global COE Program of Osaka University, the Japan Society for the Promotion of Science (JSPS), and the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- Nanobiotechnology II, ed. C. A. Mirkin and C. M. Niemeyer, Wiley-VCH, Weiheim, 2007 Search PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- P. Y. W. Dankers and E. W. Meijer, Bull. Chem. Soc. Jpn., 2007, 80, 2047–2073 CrossRef CAS.
- T. Ueno, J. Mater. Chem., 2008, 18, 3741–3745 RSC.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS.
- P. K. Lo, P. Karam, F. A. Aldaye, C. K. McLaughlin, G. D. Hamblin, G. Cosa and H. F. Sleiman, Nat. Chem., 2010, 2, 319–328 CrossRef CAS.
- P. Guo, Nat. Nanotechnol., 2010, 5, 833–842 CrossRef CAS.
- J. H. Lee, N. Y. Wong, L. H. Tan, Z. Wang and Y. Lu, J. Am. Chem. Soc., 2010, 132, 8906–8908 CrossRef CAS.
- N. Carette, H. Engelkamp, E. Akpa, S. J. Pierre, N. R. Cameron, P. C. M. Christianen, J. C. Maan, J. C. Thies, R. Weberskirch, A. E. Rowan, R. J. M. Nolte, T. M. Michon and J. C. M. van Hest, Nat. Nanotechnol., 2007, 2, 226–229 CrossRef CAS.
- Q. Wang, T. Lin, L. Tang, J. E. Johnson and M. G. Finn, Angew. Chem., Int. Ed., 2002, 41, 459–462 CrossRef CAS.
- P. A. Suci, S. Kang, M. Young and T. Douglas, J. Am. Chem. Soc., 2009, 131, 9164–9165 CrossRef CAS.
- J. C. T. Carlson, S. S. Jena, M. Flenniken, T. Chou, R. A. Siegel and C. R. Wagner, J. Am. Chem. Soc., 2006, 128, 7630–7638 CrossRef CAS; Q. Li, C. R. So, A. Fegan, V. Cody, M. Sarikaya, D. A. Vallera and C. R. Wagner, J. Am. Chem. Soc., 2010, 132, 17247–17257 CrossRef CAS.
- S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510–5514 CrossRef CAS.
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464–470 CrossRef CAS.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS.
- S. Biswas, K. Kinbara, N. Oya, N. Ishii, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2009, 131, 7556–7557 CrossRef CAS.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS.
- M. J. Boerakker, N. E. Botterhuis, P. H. H. Bomans, P. M. Frederik, E. M. Meijer, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem.–Eur. J., 2006, 12, 6071–6080 CrossRef CAS; I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2332 CrossRef CAS.
- E. N. Salgado, R. J. Radford and A. F. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS.
- K. Matsuura, K. Watanabe, T. Matsuzaki, K. Sakurai and N. Kimizuka, Angew. Chem., Int. Ed., 2010, 49, 9662–9665 CrossRef CAS.
- M. M. C. Bastings, T. F. A. de Greef, J. L. J. van Dongen, M. Merkx and E. W. Meijer, Chem. Sci., 2010, 1, 79–88 RSC.
- Strictly speaking, the term heme stands for a ferrous porphyrin. As a matter of convenience, we will employ this term throughout this paper for an iron porphyrin regardless of its oxidation state..
- H. Kitagishi, K. Oohora, H. Yamaguchi, H. Sato, T. Matsuo, A. Harada and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 10326–10327 CrossRef CAS; H. Kitagishi, K. Oohora and T. Hayashi, Biopolymers, 2009, 91, 194–200 CrossRef CAS.
- H. Kitagishi, Y. Kakikura, H. Yamaguchi, K. Oohora, A. Harada and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1271–1274 CrossRef CAS.
- M. S. Hargrove, A. J. Wilkinson and J. S. Olson, Biochemistry, 1996, 35, 11300–11309 CrossRef CAS.
- O. M. Lardinois and P. R. Ortiz de Montellano, J. Biol. Chem., 2003, 278, 36214–36226 CrossRef CAS.
- L. K. Rogers, B. L. Leinweber and C. V. Smith, Anal. Biochem., 2006, 358, 171–184 CrossRef CAS.
- F. W. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CAS.
- P. Eyer, F. Worek, D. Kiderlen, G. Sinko, A. Stuglin, V. Simeon-Rudolf and E. Reiner, Anal. Biochem., 2003, 312, 224–227 CrossRef CAS.
- One equivalent of reactive thiol in the reduced Mb(A125C) monomer was confirmed by the same method.
- K. Harada, M. Makino, H. Sugimoto, S. Hirota, T. Matsuo, Y. Shiro, Y. Hisaeda and T. Hayashi, Biochemistry, 2007, 46, 9406–9416 CrossRef CAS; B. A. Springer, S. G. Sligar, J. S. Olson and G. N. Philips, Jr., Chem. Rev., 1994, 94, 699–714 CrossRef CAS.
- U. Michelsen and C. A. Hunter, Angew. Chem., Int. Ed., 2000, 39, 764–767 CrossRef CAS.
- A. Harada, H. Yamaguchi, F. Oka and M. Kamachi, Proc. SPIE–Int. Soc. Opt. Eng., 1999, 3607, 126–135 CAS.
- E. A. Brucker, J. S. Olson, G. N. Phillips, Jr., Y. Dou and M. Ikeda-Saito, J. Biol. Chem., 1996, 271, 25419–25422 CrossRef CAS.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS.
- H. Sato, M. Watanabe, Y. Hisaeda and T. Hayashi, J. Am. Chem. Soc., 2005, 127, 56–57 CrossRef CAS.
- P. E. Mazeran, J. L. Loubet, C. Martelet and A. Theretz, Ultramicroscopy, 1995, 60, 33–40 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Synthetic and biological protocols, UV-vis spectra, ESI-TOF-MS, SDS-PAGE analyses, SEC analyses and O2 binding properties. See DOI: 10.1039/c1sc00084e/ |
| ‡ Present address: Department of Molecular Chemistry and Biochemistry, Faculty of Science and Engineering, Doshisha University, Kyotanabe, Kyoto 610-0321, Japan. |
| This journal is © The Royal Society of Chemistry 2011 |