A mononuclear nonheme iron(IV)-oxo complex which is more reactive than cytochrome P450 model compound I†
Mi Sook
Seo‡
a,
Nam Hee
Kim‡
a,
Kyung-Bin
Cho
a,
Jeong Eun
So
a,
Seon Kyung
Park
a,
Martin
Clémancey
b,
Ricardo
Garcia-Serres
b,
Jean-Marc
Latour
*b,
Sason
Shaik
*c and
Wonwoo
Nam
*a
aDepartment of Bioinspired Science, Department of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: wwnam@ewha.ac.kr; Fax: (+82) 2-32774441
bCEA/DSV/iRTSV/LCBM/pmb, CNRS UMR 5249, Université Joseph Fourier, 38054 Grenoble Cedex 09, France. E-mail: jean-marc.latour@cea.fr
cInstitute of Chemistry and The Lise Meitner-Minerva Center for Computational Quantum Chemistry, The Hebrew University of Jerusalem, 91904 Jerusalem, Israel. E-mail: sason@yfaat.ch.huji.ac.il
First published on 1st April 2011
Abstract
A highly reactive mononuclear nonheme iron(IV)-oxo complex with a low-spin (S = 1) triplet ground state in both C–H bond activation and oxo transfer reactions is reported; this nonheme iron(IV)-oxo complex is more reactive than an iron(IV)-oxo porphyrin π-cation radical (i.e., a model of cytochrome P450 compound I) and is the most reactive species in kinetic studies among nonheme iron(IV)-oxo complexes reported so far. DFT calculations support the experimental results with extremely low activation barriers in the C–H bond activation of cyclohexane and 1,4-cyclohexadiene. The DFT calculations reveal that the S = 1 state is set up to easily lead to the highly reactive S = 2 high-spin iron(IV)-oxo species.
Introduction
High-valent iron(IV)-oxo intermediates have been identified as reactive species in the catalytic cycles of dioxygen activation by mononuclear nonheme iron enzymes.1 The intermediates activate substrate C–H bonds to yield hydroxylated, desaturated, or halogenated products.2 In biomimetic studies, a number of mononuclear nonheme iron(IV)-oxo complexes have been synthesized and characterized by various spectroscopic techniques, and their chemical properties have been investigated in the oxidation of organic substrates and electron-transfer reactions.3,4 However, unlike the high-spin (S = 2) iron(IV)-oxo intermediates in nonheme iron enzymes, most of the synthetic nonheme iron(IV)-oxo complexes exhibit a low-spin (S = 1) triplet ground state.3 Therefore, much effort has been directed towards the synthesis of high-spin iron(IV)-oxo complexes, to understand the effect of the S = 2 ground spin state on the reactivities of nonheme iron(IV)-oxo intermediates in oxidation reactions.There are three examples of high-spin (S = 2) iron(IV)-oxo complexes reported in biomimetic reactions.5–7 The first high-spin iron(IV)-oxo complex, reported by Bakac and co-workers, was synthesized in the reaction of [FeII(H2O)6]2+ and ozone in acidic aqueous solution.5 Very recently, Que and co-workers reported a synthetic high-spin iron(IV)-oxo complex with a trigonal bipyramidal (TBP) geometry, [(TMG3tren)FeIV(O)]2+ (1, TMG3tren = tris(tetramethylguanidino)tren).6 Subsequently, Borovik and co-workers reported another example of a high-spin iron(IV)-oxo complex with a TBP geometry, [(H3buea)FeIV(O)]− (H3buea = tris(tert-butylureaylethylene)aminato).7 Although it has been predicted theoretically that nonheme iron(IV)-oxo species8 with a ground S = 2 spin state are more reactive than those with an S = 1 iron(IV)-oxo center due to enhancement of exchange stabilization,8a–h the high-spin iron(IV)-oxo complex reported by Que and co-workers exhibited a rather sluggish oxidizing power. In a recent computational study,8g this was shown to originate in steric encumbrance for the access of substrates to the iron-oxo moiety. Thus, it is mandatory to use an iron model without a steric hindrance8c in gauging reactivities of high-spin iron(IV)-oxo species. In addition, although it has been demonstrated that the reactivity of diiron complexes was dramatically enhanced by converting an S = 1 iron(IV)-oxo center to an S = 2 center,9 the spin state effect still remains elusive in mononuclear nonheme iron(IV)-oxo models. We therefore attempted to synthesize a mononuclear high-spin iron(IV)-oxo complex using a sterically less hindered iron(II) complex with a TBP geometry, to understand the significance of the ground S = 2 spin state of iron(IV)-oxo intermediates in nonheme iron enzymes.2 Interestingly, the iron(IV)-oxo species synthesized from a TBP iron(II) complex possessed an S = 1 ground spin state instead of an S = 2 spin state. More intriguing is the fact that this S = 1 iron(IV)-oxo complex exhibited a high reactivity capable of activating strong C–H bonds, such as those in cyclohexane (99.3 kcal mol−1), even at a low temperature (e.g., at −40 °C). To the best of our knowledge, this iron(IV)-oxo species is the most powerful oxidant in C–H bond activation and oxo transfer reactions reported so far in nonheme iron(IV)-oxo model studies.
Results and discussion
Fe(Me3NTB)(CH3CN)(CF3SO3)2 (2, Me3NTB = tris((N-methyl-benzimidazol-2-yl)methyl)amine) was synthesized by reacting equimolar amounts of FeII(CF3SO3)2(CH3CN)2 and Me3NTB (Fig. 1A) in CH3CN (see ESI, Experimental Section and Fig. S1). The crystal structure of 2 exhibits an expected TBP coordination geometry (τ = 0.86)10 (Fig. 1B; ESI, Tables S1 and S2). Addition of 1 equiv m-chloroperbenzoic acid (m-CPBA) to a solution containing 2 (1 mM) immediately gave a pale greenish yellow complex 3 with absorption bands at 380 nm (ε = ∼2700 M−1 cm−1) and 770 nm (ε = ∼200 M−1 cm−1) in CH3CN at −40 °C (Fig. 2A). The intermediate 3 was unstable and decayed fast even at −40 °C (t1/2 ∼ 2 min). Such thermal instability hampered sample preparation for spectroscopic analysis. For example, the electrospray ionization mass spectrum (ESI MS) of 3 exhibits prominent ion peaks, whose mass and isotope distribution patterns correspond to [FeIV(O)(Me3NTB)(CF3SO3)]+ and [FeIII(OH)(Me3NTB)(CF3SO3)]+ at m/z = 670.1 and 671.1, respectively (Fig. 2A, inset).![(A) Schematic drawing of Me3NTB ligand. (B) X-ray structure of [Fe(Me3NTB)(CH3CN)]2+ (see ESI, Table S2). (C) DFT calculated structure of [Fe(O)(Me3NTB)]2+ with a trigonal bipyramidal geometry (3TBP) (see ESI, Table S17). (D) DFT calculated structure of [Fe(O)(Me3NTB)(CH3CN)]2+ with an octahedral geometry (3OCT) (see ESI, Table S20). Hydrogen atoms are omitted for clarity. Calculated Fe–O distances are shown for the lowest energy structures of (S = 2) and [S = 1].](/image/article/2011/SC/c1sc00062d/c1sc00062d-f1.gif) | ||
| Fig. 1 (A) Schematic drawing of Me3NTB ligand. (B) X-ray structure of [Fe(Me3NTB)(CH3CN)]2+ (see ESI†, Table S2). (C) DFT calculated structure of [Fe(O)(Me3NTB)]2+ with a trigonal bipyramidal geometry (3TBP) (see ESI†, Table S17). (D) DFT calculated structure of [Fe(O)(Me3NTB)(CH3CN)]2+ with an octahedral geometry (3OCT) (see ESI†, Table S20). Hydrogen atoms are omitted for clarity. Calculated Fe–O distances are shown for the lowest energy structures of (S = 2) and [S = 1]. | ||
![(A) UV-vis spectrum of 3 in CH3CN at −40 °C. The inset (top) shows the observed (left) and simulated (right) ESI MS spectra of 3 with mass peaks of [FeIV(O)(Me3NTB)(CF3SO3)]+ (40%, red bars) and [FeIII(OH)(Me3NTB)(CF3SO3)]+ (60%, green bars) under the ESI MS condition. The inset (bottom) shows the magnified absorption band at 770 nm. (B) Mössbauer spectra of 3 recorded at 4.2 K (a, b, and c) or 80 K (d) under a magnetic field of 60 mT (a), 5 T (b), or 7 T (c and d) applied parallel to the γ-ray. The solid lines are simulations using the parameters given in the text. In spectra b, c, and d, the contribution of the decay product of 3 was subtracted from the experimental spectrum for the sake of clarity.](/image/article/2011/SC/c1sc00062d/c1sc00062d-f2.gif) | ||
| Fig. 2 (A) UV-vis spectrum of 3 in CH3CN at −40 °C. The inset (top) shows the observed (left) and simulated (right) ESI MS spectra of 3 with mass peaks of [FeIV(O)(Me3NTB)(CF3SO3)]+ (40%, red bars) and [FeIII(OH)(Me3NTB)(CF3SO3)]+ (60%, green bars) under the ESI MS condition. The inset (bottom) shows the magnified absorption band at 770 nm. (B) Mössbauer spectra of 3 recorded at 4.2 K (a, b, and c) or 80 K (d) under a magnetic field of 60 mT (a), 5 T (b), or 7 T (c and d) applied parallel to the γ-ray. The solid lines are simulations using the parameters given in the text. In spectra b, c, and d, the contribution of the decay product of 3 was subtracted from the experimental spectrum for the sake of clarity. | ||
The Mössbauer spectrum of 3 recorded at 4.2 K under a magnetic field of 60 mT applied parallel to γ-radiation is shown in Fig. 2B-a. The spectrum consists mostly of two quadrupole doublets in different proportions (ESI, Fig. S2 and Table S3). The most intense doublet (57% of total iron) possesses Mössbauer parameters (δ = 0.02 mm s−1, ΔEQ = 1.53 mm s−1) that match those observed for FeIV-oxo species,3b and is therefore assigned to 3. The parameters of the less intense doublet (35% of total iron; δ = 0.48 mm s−1, ΔEQ = 1.42 mm s−1) are characteristic of a (μ-oxo)diferric species, a likely decay product. As the reaction time elapses, the latter species grows at the expense of the former. Additionally, a small amount (8%) of paramagnetic species is detected at larger velocities. To firmly establish the spin state of the FeIV species, Mössbauer experiments were performed under strong magnetic fields (Fig. 2B-b, 2B-c, and 2B-d). A spin Hamiltonian simulation of the data confirmed the S = 0 ground state of the component assigned to (μ-oxo)(FeIII)2. The component assigned to 3 could be simulated only under the assumption that the spin state is S = 1. Indeed, fitting the data to an S = 2 site yields either unreasonably high D or unreasonably low A values.11 In contrast, an excellent fit to the data was obtained using parameters which are common for S = 1 iron(IV)-oxo centers (ESI, Fig. S2 and Table S4).3b D was determined to be in the range of 20–35 cm−1. The fit was performed assuming D = 28 cm−1, E/D = 0, and gx = gy = 2.3 and gz = 2.0, where g values were calculated using a method provided by Oosterhuis and Lang,12 and afforded hyperfine parameters of Ax,y,z/gNbN = (−19, −19, 0) T and η = 0.3. The component of the hyperfine coupling tensor along the main ZFS axis was undefined and was arbitrarily set equal to zero. 3 can therefore be described as an S = 1 FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. Mössbauer spectra were also recorded at temperatures as high as 160 K, and no spin transition was observed. At 180 K a sudden decrease of the Lamb–Mössbauer factor and extensive transformation of 3 made the analysis untractable.
O species. Mössbauer spectra were also recorded at temperatures as high as 160 K, and no spin transition was observed. At 180 K a sudden decrease of the Lamb–Mössbauer factor and extensive transformation of 3 made the analysis untractable.
DFT calculations were performed on 3 at B3LYP/LACV3P*+//LACVP level in order to determine its optimized structures and energies (see ESI for methodological details). First, calculations were done on a TBP geometry of 3 (3TBP, Fig. 1C), since the crystal structure of the starting complex [FeII(Me3NTB)(CH3CN)]2+ showed such a structure. The singly occupied valence electron orbital configuration of such a structure has been discussed elsewhere8a–c and consists of two-below-two degenerate orbital pairs denoted δ/δ′ and π*/π*′, respectively (Fig. 3, left side). These four orbitals are close in energy, and therefore Hund's rule is applicable in this case, resulting in high-spin occupation. The high-spin (S = 2) state was found to be 12.1 kcal mol−1 lower in energy than its low-spin (S = 1) counterpart. Interestingly, the calculations yield two electronically different configurations of the low-spin state. One of them has a valence orbital configuration, with double occupation in δ and none in δ′. This asymmetric occupancy of the δ-orbitals causes a widening of one of the Neq–Fe–Neq angles to be nearly 160°, hence the structure and electronic occupation is essentially the same as in an octahedral structure (Fig. 3, right), albeit with one equatorial ligand missing. The other S = 1 state has an electronic configuration of [π*, π*′(↓), δ, δ′], where the arrow down denotes a β-electron. This configuration is 4.5 kcal mol−1 higher in energy than the former one and is therefore least probable to form, although it retains the TBP geometry.
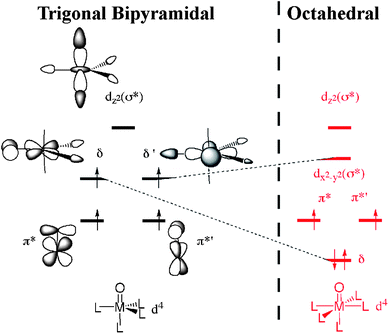 | ||
| Fig. 3 The valence electron orbitals of an ideal TBP structure (left) and of an octahedral structure (right). The four singly occupied orbitals in the TBP structure are close in energy, favouring a high-spin conformation due to favourable electron exchange interactions. Upon forming an octahedral structure (right), the degenerate δ and δ′ orbitals will split into a low and a high lying orbital, respectively. | ||
Given the Mössbauer results (vide supra), which correspond to S = 1, and that the 12.1 kcal mol−1 energy difference between the spin states is clearly out of the error margins for these calculations, the logical conclusion is that 3 is not in a TBP geometry. Hence, it is plausible that a “breathing motion” of the molecule, where an Neq–Fe–Neq angle opens up (either due to or causing a spin flip), creates a geometry that has the propensity to be stabilized by an incoming solvent molecule acting as a sixth ligand. Our calculations show that the so-formed octahedral structure (3OCT, Fig. 1D) exhibits nearly energetically degenerate spin states at room temperature, but at low temperatures the low-spin S = 1 state is preferred. This can be seen in Table 1, where removing enthalpy and entropy effects (marked in italic) would yield an energy difference of 2.0 kcal mol−1 at 0 K. As such, we conclude that the Mössbauer spectrum reflects this six-coordinate structure with a solvent acting as a sixth ligand. An alternative stereoisomer, where the oxo and solvent ligand positions are interchanged, was found to be 4.1 kcal mol−1 higher in energy and was therefore disregarded.
The oxidative reactivity of 3 was examined in the oxidation of hydrocarbons with C–H bond dissociation energies (BDE) of 77–99.3 kcal mol−1. Upon addition of substrates to a solution of 3 in CH3CN at −40 °C, the characteristic absorption band of 3 disappeared with a first-order decay profile (Fig. 4A).13 The pseudo-first-order rate constants increased proportionally with substrate concentration, from which second-order rate constants, k2, were determined (Table 2 and ESI, Fig. S4). When the k2 values were adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bonds of substrates, the k2′ values were well correlated with the BDEs of the substrates (Table 2). In addition, a deuterium kinetic isotope effect (KIE) of 26(2) was obtained when ethylbenzene-d10 was used as a substrate (Fig. 4B). Such a large KIE value implies a hydrogen tunnelling mechanism in H-atom abstraction by 3, as frequently proposed in C–H bond activation reactions by low- and high-spin iron(IV)-oxo intermediates of nonheme iron enzymes and models.3,6a,9a,14–16 Based on the observations of the correlation between reaction rates and BDEs of substrates and the large KIE value, we propose that the C–H bond cleavage of hydrocarbons by 3 is the rate-determining step in H-atom abstraction reactions.
![(A) Spectral changes observed in the reaction of [FeIV(O)(Me3NTB)]2+ (3) (1.0 mM) and ethylbenzene (EtBz, 40 mM). Inset shows absorbance traces monitored at 770 nm. (B) Plot of the pseudo-first-order rate constants, kobs (s−1), against substrate concentrations to determine second-order rate constants, k2, and C–H kinetic isotope effect (KIE) value for the reaction of [FeIV(O)(Me3NTB)]2+ (3) with ethylbenzene (■, black solid line, k2 = 1.49 M−1s−1) and deuterated ethylbenzene-d10 (●, red solid line, k2 = 5.7 × 10−2 M−1s−1) in CH3CN at −40 °C.](/image/article/2011/SC/c1sc00062d/c1sc00062d-f4.gif) | ||
| Fig. 4 (A) Spectral changes observed in the reaction of [FeIV(O)(Me3NTB)]2+ (3) (1.0 mM) and ethylbenzene (EtBz, 40 mM). Inset shows absorbance traces monitored at 770 nm. (B) Plot of the pseudo-first-order rate constants, kobs (s−1), against substrate concentrations to determine second-order rate constants, k2, and C–H kinetic isotope effect (KIE) value for the reaction of [FeIV(O)(Me3NTB)]2+ (3) with ethylbenzene (■, black solid line, k2 = 1.49 M−1s−1) and deuterated ethylbenzene-d10 (●, red solid line, k2 = 5.7 × 10−2 M−1s−1) in CH3CN at −40 °C. | ||
| Substrate | BDE (kcal mol−1)b | k 2/M−1s−1 | k 2′/M−1s−1 |
|---|---|---|---|
| a Second-order rate constants, k2, were determined at −40 °C and adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bond of substrates. Products from desaturation and hydroxylation reactions were obtained, and the results of product analysis, including DFT calculations, will be communicated in a separate paper. Also, see Product Analysis in ESI, Experimental Section. b BDE of C–H bonds are from ref. 15 and 16 in the text. | |||
| 9,10-Dihydroanthracene | 77 | 3.1 × 103 | 7.8 × 102 |
| 1,4-Cyclohexadiene | 78 | 9.4 × 102 | 2.4 × 102 |
| Triphenylmethane | 81 | 1 × 101 | 1 × 101 |
| Cumene | 85 | 1.0 | 1.0 |
| Ethylbenzene | 87 | 1.5 | 7.5 × 10−1 |
| Toluene | 90 | 4.7 × 10−1 | 1.6 × 10−1 |
| Cyclooctane | 95.3 | 2.2 | 1.4 × 10−1 |
| 2,3-Dimethylbutane | 96.5 | 2.9 × 10−1 | 1.5 × 10−1 |
| Cyclohexane | 99.3 | 2.5 × 10−1 | 2.0 × 10−2 |
The reactivity of 3 was then compared to those of iron(IV)-oxo nonheme mononuclear, dinuclear and porphyrin π-cation radical complexes (i.e., comparisons of low-spin vs. high-spin states of mononuclear and dinuclear nonheme irons and nonheme vs.heme iron models). In the reactivity comparisons, we found that 3 is the most powerful oxidant in C–H bond activation and oxo transfer reactions (see data in Table 3). In the oxidation of 1,4-cyclohexadiene (CHD) and 9,10-dihydroanthracene (DHA) (i.e., C–H bond activation reactions), 3 was the most reactive of mononuclear nonheme iron(IV)-oxo species (Table 3, column of C–H bond activation; also see ESI, Fig. S4); the reactivity of 3 was >1800 times greater than that of a highly reactive iron(IV)-oxo species, [(N4Py)FeIVO]2+ (low-spin iron(IV)-oxo species 4, N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine).16,17 Similarly, 3 was 2600 times more reactive than [(Bn-tpen)FeIVO]2+ (low-spin iron(IV)-oxo species 5, Bn-tpen = N-benzyl-N,N′,N′-tris(2-pyridylmethyl)ethane-1,2-diamine) in the oxidation of ethylbenzene (k2 = 1.5 and 5.8 × 10−4 M−1s−1 at −40 °C for 3 and 5, respectively; see ESI, Fig. S5); 5 is a strong oxidant that can oxidize C–H bonds of cyclohexane (99.3 kcal mol−1) at room temperature.16 Interestingly, the oxidizing power of 3 was comparable to that of a highly reactive diiron complex with a high-spin [HO–(L)FeIII–O–FeIV(L)O]2+ core (6)9a (compare k2 values in the oxidation of DHA by 3 and 6 in Table 3, column of DHA). More interestingly, when the reactivities of 3 and cytochrome P450 model compound I were compared in the C–H bond activation of alkanes, 3 showed a greater reactivity than [(TDCPP)+˙FeIVO]+ (high-spin iron(IV)-oxo porphyrin π-cation radical 7, TDCPP = meso-tetrakis(2,6-dichlorophenyl)porphinato dianion) (Fig. 5; see ESI, Fig. S6 for the determination of k2 values of 7). Recalling that 7 bearing an electron-deficient porphyrin ligand is a highly reactive oxidant in alkane hydroxylation reactions,18 one can further infer that 3 is the most powerful oxidant in C–H bond activation of hydrocarbons. Activation parameters for the oxidation reaction of 3 with 1,4-cyclohexadiene (CHD), determined by Eyring analysis (233–243 K) are ΔH‡ = 10(1) kcal mol−1 and ΔS‡ = −1.0 cal mol−1 K−1 (see Fig. S7).
| FeIV(O) | C–H bond activationk2 [M−1s−1] | Oxo transfer k2 [M−1s−1] | ||
|---|---|---|---|---|
| CHD | DHA | PhSMe | PPh3 | |
| a Data from ref. 6. b Data from this work. c Not determined due to fast reaction. d Data from ref. 9a. e Substrate was P(C6H5)2(C6F5). | ||||
| 1 | 1.2 (−30 °C)a | 1.3 (−30 °C)a | ||
| 3 | 9.4 × 102 (−40 °C)b | 3.1 × 103 (−40 °C)b | 2.1 × 104 (−40 °C)b | NDc |
| 4 | 5.0 × 10−1 (−40 °C)b | 8.0 × 10−1 (−40 °C)b | 2.4 × 10−4 (−40 °C)b | 1.5 (−30 °C)a |
| 5 | 5.7 (−40 °C)b | 8.8 (–40 °C)b | 1.4 × 10−2 (−40 °C)b | 1.7 × 10 (−40 °C)b |
| 6 | 2.8 × 10 (−80 °C)d | 2.0 (−80 °C)d,e | ||
![Plot of log k2′ of [FeIV(O)(Me3NTB)]2+ (3, black filled squares), and [(TDCPP)+˙FeIVO]+ (7, red filled circles) against C–H BDE of substrates. Second-order rate constants, k2, were determined at −40 °C and then adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bonds of substrates. See ESI, Fig. S6 for the determination of k2′ values of 3 and 7.](/image/article/2011/SC/c1sc00062d/c1sc00062d-f5.gif) | ||
Fig. 5 Plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k2′ of [FeIV(O)(Me3NTB)]2+ (3, black filled squares), and [(TDCPP)+˙FeIVO]+ (7, red filled circles) against C–H BDE of substrates. Second-order rate constants, k2, were determined at −40 °C and then adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bonds of substrates. See ESI, Fig. S6 for the determination of k2′ values of 3 and 7. k2′ of [FeIV(O)(Me3NTB)]2+ (3, black filled squares), and [(TDCPP)+˙FeIVO]+ (7, red filled circles) against C–H BDE of substrates. Second-order rate constants, k2, were determined at −40 °C and then adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bonds of substrates. See ESI, Fig. S6 for the determination of k2′ values of 3 and 7. | ||
Reactivities of the nonheme iron(IV)-oxo complexes were also compared in oxo transfer reactions. First, the reactivity of 3 could not be compared to other iron(IV)-oxo complexes in the oxidation of PPh3 due to its too fast reaction; it was impossible to determine the reaction rate even with a stopped-flow spectrometer (Table 3, column of PPh3). Although kinetic data were not available for 1 and 6 in the oxidation of thioanisole (PhSMe) (Table 3, column of PhSMe), comparing the kinetic data in the oxidation of PhSMe and PPh3 leads us to deduce that 3 is the most reactive oxidant in oxo transfer reactions by nonheme iron(IV)-oxo species, including the mononuclear and dinuclear high-spin iron(IV)-oxo complexes (i.e., 1 and 6). For example, the reactivity of 4 was comparable to that of 1 and slightly lower than that of 6 in the oxidation of PPh3 (Table 3, column of PPh3). However, 3 was 108-fold more reactive than 4 in the oxidation of PhSMe (compare data in italic in Table 3)! Based on the reactivity comparisons, we conclude that 3 is the most powerful oxidant in oxo transfer reactions among the mononuclear and dinuclear nonheme iron(IV)-oxo complexes reported so far.
The reactant state assignment as S = 1 would seemingly imply that the above results prove that S = 1 is the reactive state and hence, the spin state identity of nonheme iron(IV)-oxo complexes does not play an important role in determining reactivities in both the C–H bond cleavage and the oxo transfer reactions. Indeed, using ground state spin assignments on RuIVO complexes, there have been experimental suggestions that the spin states do not matter for reactivities.19 However, since all calculations,8 including some experimental data,9a show that the S = 2 state is by far the most reactive one, this notion must be carefully examined. Since the DFT calculations showed that the S = 1 and S = 2 spin states are very close in energy for the six-coordinated reagent (3OCT), the S = 2 state may in fact be easily accessible and be the one that mediates the reactivity. We therefore studied the reactivities of 3TBP and 3OCT towards C–H bond activation of cyclohexane as well as CHD.
The geometries of the two transition states of the high-spin 3 are shown in Fig. 6 for the case of cyclohexane. As discussed elsewhere,8a–c,g–k there is a close relationship between the interacting geometries and the different spin-states. The high-spin state involves an α-electron donation from the substrate to the σ*z2 orbital during the C–H activation reaction. This can be seen in the spin density distribution during the reaction, where the substrate develops a β-radical (Table 4). The overlap between σ*z2 and the substrate is maximized if the angle Fe–O–H is linear; hence the high-spin transition states in Fig. 6 both show this feature. The low-spin state, on the other hand, involves a transfer of a β-electron from the substrate, as seen in Table 4 where the substrates develop an α-radical. The receiving orbital is in this case one of the π* orbitals, hence maximal overlap is obtained if the substrate interacts with the oxygen side-on. Therefore, the low-spin transition states here show a markedly bent angle (ESI, Tables S21 and S22).
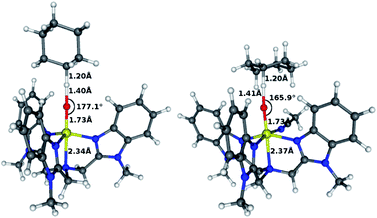 | ||
| Fig. 6 High-spin trigonal bipyramidal (left) and octahedral (right) transition states in C–H activation of cyclohexane. | ||
| FeO | 5 × ligated N | Substrate | |
|---|---|---|---|
| a See also ESI Tables S15 and S16. | |||
| S = 1 | |||
| Reactants | 2.09/2.08 | −0.03/−0.04 | 0.00/0.00 |
| Transition state | 1.55/1.66 | −0.02/−0.01 | 0.49/0.38 |
| Intermediate | 1.10/1.08 | −0.03/−0.05 | 0.95/0.99 |
| S = 2 | |||
| Reactants | 3.82/3.81 | 0.17/0.16 | 0.00/0.00 |
| Transition state | 4.00/3.92 | 0.29/0.25 | −0.31/−0.19 |
| Intermediate | 4.59/4.64 | 0.31/0.28 | −0.92/−0.96 |
Fig. 7 summarizes the calculation results for both the substrates. In the far left section, the high-spin state of 3TBP is shown to be 12.1 kcal mol−1 more stable than the low-spin state (see also ESI, Table S5). Binding of a solvent molecule, in the second section, will turn 3TBP into 3OCT. As seen by the blue and black lines, this ligation is slightly exothermic and causes the spin states to become degenerate at room temperature. As argued already above, at lower temperatures, at which the Mössbauer experiments were done, entropy and enthalpy effects slightly favor the low-spin state (ESI, Table S8). When the substrate is added (the third section, “+ cyclohexane” or “+ CHD”), the reaction can easily switch to the S = 2 state and mediate the H-abstraction reaction with a small barrier (4.9 kcal mol−1 relative to 3OCT for cyclohexane, ESI Table S10, and 0.3 kcal mol−1 relative to the reactant complex for CHD, ESI Table S9). These calculated barriers are lower than those that would be extracted from the k2 values in Table 3. This is understandable because the computed barriers do not include all factors which may affect the experimental free energy barriers, for instance, translation and rotational loss of entropy due to complexation, and the effect of counterions, since these effects cannot be easily calculated. However, these computed values do indeed indicate that this particular iron-oxo complex is very reactive, and comparing to the calculated S = 1 path (wherein the barrier is 15.7/7.7 kcal mol−1 for both substrates, respectively), the high-spin path is clearly preferred. Fig. 7 displays also the energy profile for C–H activation by 3TBP directly in S = 2 (e.g. without a solvent ligation, red dashed lines), and one can see here too a small activation barrier of 5.7/2.9 kcal mol−1.
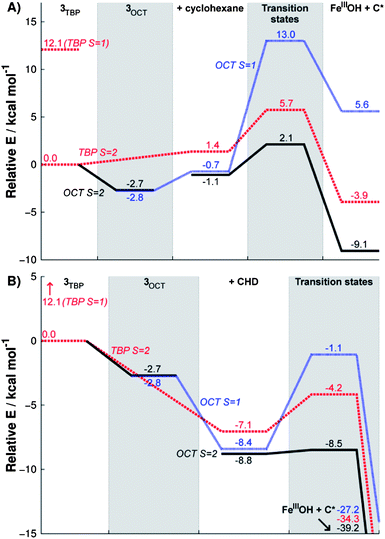 | ||
| Fig. 7 Reaction energy profiles of C–H activation reaction of cyclohexane (A) and CHD (B) by 3OCT in high-spin (black, solid) and low-spin (blue, dotted) and by 3TBP in high-spin (red, dashed). The high energy low-spin state 3TBP is also indicated. | ||
Thus, the theoretical calculations predict mainly two possible scenarios. One scenario involves initially 3OCT in its octahedral geometry with a low-spin ground state. A spin flip then occurs to give the high-spin state which mediates the reaction via a low-lying transition state, whose geometry is shown in the right hand side of Fig. 6 in the case of cyclohexane. The other scenario is that the six-coordinated species liberates its sixth coordinated solvent ligand [eqn (1)] and converts to the high-spin TBP geometry (3TBP) that reacts with the substrate (Fig. 6, left). These options predict that the reactive spin state is indeed the high-spin state.
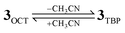 | (1) |
Conclusions
We have reported a highly reactive mononuclear nonheme iron(IV)-oxo complex in both C–H bond activation and oxo transfer reactions with a reactant low-spin (S = 1) triplet ground state; this intermediate is more reactive than an iron(IV)-oxo porphyrin π-cation radical (i.e., a model of cytochrome P450 compound I) and is the most reactive species among nonheme iron(IV)-oxo complexes reported so far. Results from DFT calculations are consistent with this view as the calculated activation barriers of the C–H bond activations are extremely low with both cyclohexane and 1,4-cyclohexadiene. However, DFT calculations show that the reactive state is most likely the closely-lying S = 2 state, as predicted by many computational data8 and a recent experimental result.9a The observation of such a high reactivity coming from a nonheme iron(IV)-oxo complex with a reactant S = 1 ground spin state shows that inferring from the nature of the ground state on reactivity is not safe, since the S = 2 spin state may well be very low lying and its exchange-enhanced reactivity8a–c,g,o mediates the H-abstraction reactivity at much lower barriers than the S = 1 spin state. Thus, the complex 3 described herein is in a stable S = 1 reactant state with a highly reactive S = 2 spin state. This is probably the reason why Nature has selected nonheme enzymes with an S = 2 spin state.Acknowledgements
The research at EWU was supported by NRF/MEST of Korea through the CRI, GRL (2010-00353), WCU (R31-2008-000-10010-0), and Basic Research Program (2010-0002558 to MSS). JML acknowledges the support of the Region Rhone-Alpes through contract CIBLE 07 016335. SS thanks the Israel Science Foundation (Grant 53/09).Notes and references
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS , and review articles in the special issue; (b) P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716 RSC; (c) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227 CrossRef CAS; (d) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr., Chem. Rev., 2004, 104, 939 CrossRef CAS.
- (a) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger, Jr., Acc. Chem. Res., 2007, 40, 484 CrossRef CAS; (b) J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger, Jr. and C. Krebs, Biochemistry, 2003, 42, 7497 CrossRef CAS; (c) L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger, Jr. and C. Krebs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738 CrossRef CAS; (d) D. P. Galonić, E. W. Barr, C. T. Walsh, J. M. Bollinger, Jr. and C. Krebs, Nat. Chem. Biol., 2007, 3, 113 CrossRef CAS; (e) B. E. Eser, E. W. Barr, P. A. Frantom, L. Saleh, J. M. Bollinger, Jr., C. Krebs and P. F. Fitzpatrick, J. Am. Chem. Soc., 2007, 129, 11334 CrossRef CAS; (f) D. A. Proshlyakov, T. F. Henshaw, G. R. Monterosso, M. J. Ryle and R. P. Hausinger, J. Am. Chem. Soc., 2004, 126, 1022 CrossRef CAS.
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS; (b) L. Que, Jr., Acc. Chem. Res., 2007, 40, 493 CrossRef CAS.
- (a) Y.-M. Lee, H. Kotani, T. Suenobu, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 434 CrossRef CAS; (b) S. Fukuzumi, H. Kotani, T. Suenobu, S. Hong, Y.-M. Lee and W. Nam, Chem.–Eur. J., 2010, 16, 354 CrossRef CAS; (c) P. Comba, S. Fukuzumi, H. Kotani and S. Wunderlich, Angew. Chem., Int. Ed., 2010, 49, 2622 CrossRef CAS; (d) S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Nat. Chem., 2010, 2, 756 CrossRef CAS; (e) Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 403 CrossRef CAS; (f) J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Jr., Science, 2003, 299, 1037 CrossRef CAS; (g) M. S. Seo, J.-H. In, S. O. Kim, N. Y. Oh, J. Hong, J. Kim, L. Que, Jr. and W. Nam, Angew. Chem., Int. Ed., 2004, 43, 2417 CrossRef CAS; (h) J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257 CrossRef CAS; (i) M. Martinho, F. Banse, J.-F. Bartoli, T. A. Mattioli, P. Battioni, O. Horner, S. Bourcier and J.-J. Girerd, Inorg. Chem., 2005, 44, 9592 CrossRef CAS; (j) A. Thibon, J.-F. Bartoli, S. Bourcier and F. Banse, Dalton Trans., 2009, 9587 RSC.
- (a) O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Münck, L. Que, Jr. and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871 CrossRef CAS; (b) O. Pestovsky and A. Bakac, J. Am. Chem. Soc., 2004, 126, 13757 CrossRef CAS.
- (a) J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck and L. Que, Jr., Angew. Chem., Int. Ed., 2009, 48, 3622 CrossRef CAS; (b) J. England, Y. Guo, E. R. Farquhar, V. G. Young, Jr., E. Münck and L. Que, Jr., J. Am. Chem. Soc., 2010, 132, 8635 CrossRef CAS.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188 CrossRef CAS.
- (a) H. Hirao, L. Que, Jr., W. Nam and S. Shaik, Chem.–Eur. J., 2008, 14, 1740 CrossRef CAS; (b) H. Hirao, D. Kumar, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590 CrossRef CAS; (c) K.-B. Cho, S. Shaik and W. Nam, Chem. Commun., 2010, 46, 4511 RSC; (d) D. Kumar, H. Hirao, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026 CrossRef CAS; (e) H. Hirao, D. Kumar, W. Thiel and S. Shaik, J. Am. Chem. Soc., 2005, 127, 13007 CrossRef CAS; (f) L. Bernasconi, M. J. Louwerse and E. J. Baerends, Eur. J. Inorg. Chem., 2007, 3023 CrossRef CAS; (g) D. Janardanan, Y. Wang, P. Schyman, L. Que, Jr. and S. Shaik, Angew. Chem., Int. Ed., 2010, 49, 3342 CAS; (h) H. Chen, W. Lai and S. Shaik, J. Phys. Chem. Lett., 2010, 1, 1533 Search PubMed; (i) C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717 CrossRef CAS; (j) S. Ye and F. Neese, Curr. Opin. Chem. Biol., 2009, 13, 89 CrossRef CAS; (k) E. Godfrey, C. S. Porro and S. P. de Visser, J. Phys. Chem. A, 2008, 112, 2464 CrossRef CAS; (l) S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632 CrossRef CAS; (m) L. Bernasconi, M. J. Louwerse and E. J. Baerends, Eur. J. Inorg. Chem., 2007, 3023 CrossRef CAS; (n) S. P. de Visser, R. Latifi, L. Tahsini and W. Nam, Chem.–Asian J., 2011, 6, 493 CrossRef CAS; (o) S. Shaik, H. Chen and D. Janardanan, Nat. Chem., 2011, 3, 19 CrossRef CAS.
- (a) G. Xue, R. De Hont, E. Münck and L. Que, Jr., Nat. Chem., 2010, 2, 400 CrossRef CAS; (b) S. Shaik, Nat. Chem., 2010, 2, 347 CrossRef CAS; (c) D. Wang, E. R. Farquhar, A. Stubna, E. Münck and L. Que, Jr., Nat. Chem., 2009, 1, 145 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- M. H. Lim, J.-U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que, Jr., Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665 CrossRef CAS.
- W. T. Oosterhuis and G. Lang, J. Chem. Phys., 1973, 58, 4757 CrossRef CAS.
- Reaction solutions of in situ generated 3 were pure enough to be used for kinetic studies. Experimental details for product analysis are described in ESI, Experimental Section.
- J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger, Jr., J. Am. Chem. Soc., 2003, 125, 13008 CrossRef CAS.
- (a) C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, Jr., S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181 CrossRef; (b) J. P. Roth and J. M. Mayer, Inorg. Chem., 1999, 38, 2760 CrossRef CAS; (c) J. P. Mayer, Biomimetic Oxidations Catalyzed by Transition Metal Complexes, ed. B. Meunier, Imperial College Press, London, 2000, pp. 1–43 Search PubMed.
- (a) J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, Jr., J. Am. Chem. Soc., 2004, 126, 472 CrossRef CAS; (b) J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger, Jr. and C. Krebs, Biochemistry, 2003, 42, 7497 CrossRef CAS; (c) J. C. Price, E. W. Barr, B. Tirupati, C. Krebs and J. M. Bollinger, Jr., J. Am. Chem. Soc., 2003, 125, 13008 CrossRef CAS; (d) J. T. Groves and Y.-Z. Han, in Cytochrome P450: Structure, Mechanism and Biochemistry, 2nd edn, ed. P. R. Ortiz de Montellano, Plenum Press, New York, pp. 3–48 Search PubMed.
- C. V. Sastri, K. Oh, Y. J. Lee, M. S. Seo, W. Shin and W. Nam, Angew. Chem., Int. Ed., 2006, 45, 3992 CrossRef CAS.
- (a) D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251 CrossRef CAS; (b) Y. M. Goh and W. Nam, Inorg. Chem., 1999, 38, 914 CrossRef CAS; (c) S. R. Bell and J. T. Groves, J. Am. Chem. Soc., 2009, 131, 9640 CrossRef CAS.
- T. Kojima, Y. Hirai, T. Ishizuka, Y. Shiota, K. Yoshizawa, K. Ikemura, T. Ogura and S. Fukuzumi, Angew. Chem., Int. Ed., 2010, 49, 8449 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectroscopic, computational and kinetic details. See DOI: 10.1039/c1sc00062d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |