exTTF-capped gold nanoparticles as multivalent receptors for C60†
Roberto
Cao
Jr.
ab,
Helena
Isla
b,
Roberto
Cao
a,
Emilio M.
Pérez
bc and
Nazario
Martín
*bc
aLaboratorio de Bioinorgánica, Facultad de Química Universidad de LaHabana, La Habana, 10400, Cuba
bDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040, Madrid, Spain. E-mail: nazmar@quim.ucm.es; Fax: +34-91-394-4103
cIMDEA Nanociencia, Facultad de Ciencias, Avda. Fco. Tomás y Valiente, 7, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
First published on 13th May 2011
Abstract
We report the synthesis and C60 binding abilities of gold nanoparticles capped with π-extended tetrathiafulvalene (exTTFAuNPs). The exTTFAuNPs show a strong tendency to aggregate both in solution and in the solid state through multiple weak exTTF-exTTF interactions. A thorough collection of experiments demonstrates that upon addition of C60, stable noncovalent exTTFAuNP·C60 associates are formed, with concomitant partial disaggregation of the nanoparticles.
Introduction
Framed within the search for new materials for energy conversion, there is a growing interest in the exploration of noncovalent associates of electroactive molecules.1,2 With regards to electron donors, the supramolecular properties of tetrathiafulvalene have been thoroughly explored,3 while the noncovalent chemistry of its π-extended analogue 9,10-di(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (exTTF) had hardly been investigated. Building on the design of the first receptors for fullerene based on exTTF,4 our group has contributed to this area with the investigation of the supramolecular associates between π-extended tetrathiafulvalenes and fullerenes.4–6 Thus far, we have reported the construction of linear6a and hyperbranched6b supramolecular polymers, covalent dendrimers capable of associating multiple units of fullerene,6c and fully electron acceptor variants of the former two.6d Based on the example of the covalent dendrimers, we reasoned we could increase the number of fullerene binding sites and significantly reduce the synthetic effort by utilizing gold nanoparticles as scaffolds. This approach has been successfully utilized in the construction of multivalent receptors for numerous analytes.7 Moreover, the attachment of electroactive molecules to gold nanoparticles has received considerable attention, especially in the case of porphyrins,8 but to date, TTF and its derivatives have played a secondary role.9Here we present the synthesis, characterization, and supramolecular properties of gold nanoparticles densely functionalised with exTTF.
Results and discussion
It has been shown that gold nanoparticles (AuNPs) functionalized with porphyrins through alkanethiole spacers bind fullerenes in solution, with the AuNP acting as a linker between the two necessary porphyrin units.8f Taking this into account, we designed and synthesised exTTF-capped AuNPs (exTTFAuNPs) according to Scheme 1. Firstly, we prepared AuNPs by reduction of HAuCl4 with sodium citrate in water, following the classic protocol described by Frens.10 Separately, we prepared the exTTF ester of mercaptoundecanoic acid (MUA-exTTF) by condensation of readily available exTTF-CH2OH and MUA in CH2Cl2 with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride as coupling reagent and 4-dimethylaminopyridine as base and catalyst. To obtain the exTTFAuNPs we performed a liquid–liquid extraction, with the MUA-exTTF dissolved in chlorobenzene as organic phase and the citrate-capped AuNPs in aqueous solution. This mixture was stirred vigorously for two hours or until complete decoloration of the red aqueous phase. The golden-green organic phase was centrifuged with CH2Cl2 to precipitate the exTTFAuNPs, which were further washed several times with CH2Cl2 and CH3OH to remove excess organic ligand (for experimental details, see the Supporting Information†). The exTTFAuNPs thus obtained show a diameter of 7.4 ± 1.0 nm, as calculated from statistical analysis of transmission electron microscopy (TEM) images (Figure S1†).![Synthesis of exTTFAuNPs and representation of their aggregation and partial disaggregation upon binding of [60]fullerene.](/image/article/2011/SC/c1sc00179e/c1sc00179e-s1.gif) | ||
| Scheme 1 Synthesis of exTTFAuNPs and representation of their aggregation and partial disaggregation upon binding of [60]fullerene. | ||
The electronic absorption spectrum of the exTTFAuNPs (chlorobenzene, 298 K) shows a band at λ ∼ 450 nm in a region in which the spectrum of MUA-AuNPs shows a minimum (Figure S2†). We attribute this absorption to the exTTF chromophore (λmax = 434 nm). Besides this, an intense and broad absorption band at λ ∼ 590 nm, characteristic of AuNPs forming aggregates11 (Fig. 1) is also found. We had previously observed the same tendency to self-associate in the case of covalent dendrimers featuring numerous exTTF units in their outer shell.6c In the case of the dendrimers, upon addition of C60 the weak exTTF-exTTF interactions were readily substituted with the energetically more favorable exTTF-C60,4–6,12 which allowed for the recognition of multiple units of C60. This is also the case for the exTTFAuNPs, as shown in Fig. 1. Upon addition of C60 (chlorobenzene, 0.52 mM), we observed depletion of the band at λ ∼ 450 nm, which is the signature of the exTTF·C60 recognition event.4,5 Besides this, we observed a decrease in intensity and a slight hypsochromic shift in the plasmon resonance band (λ ∼ 590 nm), indicating partial disaggregation of the nanoparticles. In order to confirm this last observation a computational simulation was carried out (see the Supporting information†).13 Briefly, the optical response of aggregated nanoparticles was described using a simplified model composed of three aligned gold nanoparticles in which one of them was set to increase its distance from the other two. Our computational model confirms that an increase in the distance between the nanoparticles leads to a decrease in the intensity and a blue shift of the plasmon band (Figure S3†), as observed experimentally.
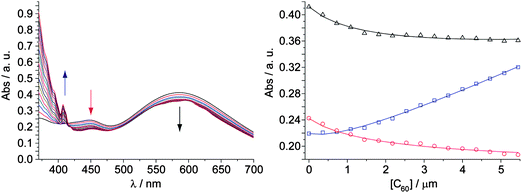 | ||
Fig. 1 Left: changes in the UV-vis spectrum (chlorobenzene, 298 K) of exTTFAuNPs with increasing concentration of C60. Right: binding isotherms for a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exTTF:C60 model at 406 nm (blue), 449 nm (red) and 587 nm (black); empty squares, circles and triangles are experimental data, solid lines correspond to the fit for a binding constant of log β5:1 = 22.5 ± 0.1. :1 exTTF:C60 model at 406 nm (blue), 449 nm (red) and 587 nm (black); empty squares, circles and triangles are experimental data, solid lines correspond to the fit for a binding constant of log β5:1 = 22.5 ± 0.1. | ||
The absence of tight isosbestic points is in agreement with the coexistence of several species of different stoichiometry during the titration, as was expected. The quantitative analysis of such complex titration data is not straightforward. Firstly, we calculated the molar concentration of exTTF chromophores in the solution of the nanoparticles considering the absorbance at 450 nm and the molar absorptivity of exTTF. Secondly, in order to get an idea of the possible exTTF:C60 stoichiometry of the associates, we carried out continuous variation plots, which resulted in several poorly defined maxima. This disappointing result is due to the tendency of Job's plots to fail when more than one type of complexes are present in solution.14 Thus, we resorted to molecular mechanics calculations (AMBER),15 which indicated that up to five units of MUA-exTTF can surround C60 without steric congestion (Fig. 2). Indeed, the UV-vis titration data (Fig. 1) fitted successfully to either a 5:1 or a 4:1 exTTF:C60 binding models utilizing global multivariable analysis software, while fitting to other reasonable models (2:1, 3:1, and 6:1) was unsuccessful (Figure S4†). The analysis yielded binding constants of log β5:1 = 22.5 ± 0.1 or log β4:1 = 17.9 ± 0.1. Assuming all exTTF-C60 binding events present the same binding constant, both of these values correspond to a log K1:1 = 4.5, indicative of a very strong multivalent effect. For comparison, our original tweezers-like hosts show a binding constant one order of magnitude smaller, despite featuring two units of exTTF (log Ka = 3.5).4c Only highly preorganized macrocyclic bis-exTTF receptors show binding constants comparable to the exTTFAuNPs (log Ka 4–6.5).5b In fact, we have not observed spectroscopic evidence of binding C60 for a single unit of exTTF in the absence of the gold nanoparticle scaffold,4c unless other cooperative forces are also present.5c,f
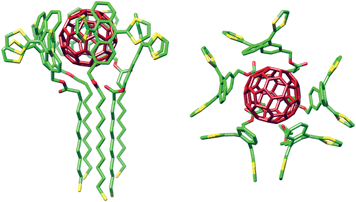 | ||
| Fig. 2 Side and top views of the energy-minimized (AMBER) model of the associate with five units of MUA-exTTF surrounding C60. Carbon atoms are depicted in green, sulfurs in yellow and oxygens in red. Hydrogens are omitted for clarity. C60 is depicted in dark red. | ||
Fourier transform infrared spectroscopy (FTIR) of samples of exTTFAuNPs and exTTFAuNP·C60 also provided evidence of complexation/dissaggregation. Most characteristic signals of the exTTF fragment are significantly shifted (8–4 cm−1) upon complexation, and a remarkable sharpening of the signal corresponding to the substituted benzene ring (735 cm−1) is also observed. Meanwhile the F1u(3) and F1u(4) modes of C60 in the exTTFAuNP·C60 appear at 1180 and 1428 cm−1, respectively (Figure S5†), in good agreement with data reported for complexes of C60 with TTF-type donors.16
Exploration under atomic force microscopy (AFM, tapping mode, air) confirmed the proposed scenario (Fig. 3). Images of a dropcast of a saturated solution of the exTTFAuNPs in 1,2-dichlorobenzene on freshly exfoliated mica, showed exclusively large (ca. 0.5–1μm) clusters of nanoparticles of approximately 100 nm in height (i.e. 10–15 exTTFAuNP layers) separated by large void areas. Neither smaller clusters nor single exTTFAuNPs were observed. When the same solution was incubated with C60 for two hours before deposition, AFM revealed large areas of fullerene precipitate (1–5 nm, Fig. 3c and d), together with clusters of nanoparticles of 50–100 nm in height (i, Fig. 3c and d), and abundant smaller aggregates of exTTFAuNP·C60 comprising approximately 2–3 layers of nanoparticles (15–25 nm, ii, Fig. 3c and d).
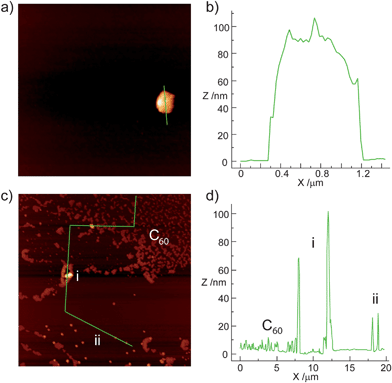 | ||
| Fig. 3 AFM imaging of a) dropcast of a saturated solution of exTTFAuNPs in 1,2-dichlorobenzene, showing a cluster of nanoparticles and c) dropcast of a saturated solution of exTTFAuNPs incubated with C60 for 2h prior to dropcasting. b) and d) show profiles along the green lines in a) and c), respectively. | ||
TEM imaging was also consistent with the UV-vis data and AFM pictures (Fig. 4). Pristine exTTFAuNPs are heavily aggregated, forming large clusters of nanoparticles in very close contact, as shown in Fig. 4a. After incubation with C60, however, the size of the clusters is reduced, and the relative distance between the nanoparticles increased, to the point that some single exTTFAuNP·C60 are observed (Fig. 4c).
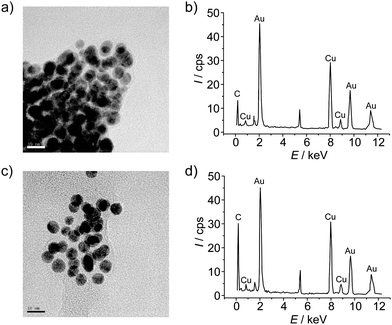 | ||
| Fig. 4 a) TEM micrograph of exTTFAuNPs and b) its EDX spectrum. c) TEM micrograph of exTTFAuNP·C60 and d) its EDX spectrum. Scale bars on the micrographs represent 10 nm. | ||
Energy dispersive X-ray spectroscopy (EDX) of the TEM micrographs is also in agreement with the binding event. In the case of pristine exTTFAuNPs (Fig. 4b), besides the signature signals of the copper grid, we observed peaks at 2.2, 9.7 and 11.5 keV, corresponding to gold, and at 0.2 keV, corresponding to carbon. The sulfur peak is most likely too weak to be detected or overlapped with the intense Au peak at 2.2 keV. As for the exTTFAuNP·C60spectrum, the main features are identical, but the relative intensity of the carbon signal compared to the most prominent gold peak is approximately 0.7, compared to 0.3 in exTTFAuNPs, in accordance with a high loading of C60.
Finally, we have also carried out cyclic voltammetry (CV) measurements of exTTFAuNPs, exTTFAuNP·C60 and C60 to investigate the influence of the binding event on the electronic properties of the electroactive units. Measurements were carried out in chlorobenzene containing 0.1 M Bu4NPF6 under argon atmosphere. A glassy carbon was used as working electrode, Ag/AgNO3 as the reference electrode, and a platinum wire as the counter. Ferrocene (Fc) was added as an internal reference. Table 1 shows the results of the CV experiments.
The CV of exTTFAuNPs shows a single oxidation process at a peak potential of 0.386 V, with marked absorption and desorption processes dominating the anodic part of the voltammogram and preventing determination of the back-wave potential. Upon addition of C60, the oxidation wave is significantly broadened, and three quasi-reversible reduction processes involving the carbon cage are detected. These reductions appear at half-wave potentials of −1.183, −1.584, and −2.059 V. For comparison, the cyclic voltammetry of C60 under identical experimental conditions showed similar reduction processes, although at less negative potential values. Typically, the reductions show a cathodic shift of ∼ 30 mV in exTTFAuNP·C60 compared to C60 (see Table 1 and Figure S6†). Remarkably, all three waves are affected to a similar extent, indicating that the fullerene unit remains associated upon the first and second reduction processes. These results are in line with those we have reported previously for other exTTF-based hosts for fullerenes.4,5
Conclusions
In summary, we have presented the synthesis and fullerene binding abilities of the first exTTF-capped gold nanoparticles. The synthesis of the exTTFAuNPs was carried out following standard and preparatively straightforward protocols, both for the gold nanoparticles and the organic addend. The exTTFAuNPs are heavily aggregated through multiple weak van der Waals interactions between the exTTF units, as shown by UV-vis, AFM and TEM. These weak exTTF-exTTF interactions are readily substituted in solution with the stronger exTTF-C60. Indeed, a thorough collection of experiments demonstrates that the exTTFAuNPs are capable of associating C60 both in solution (UV-vis, CV) and in the solid state (FTIR, AFM, TEM, EDX), and that binding occurs with concomitant partial disaggregation of the nanoparticles. Quantitative analysis of the UV-vis titration data was performed utilizing global multivariable analysis software, and shows that the exTTFAuNP·C60 associates are extremely stable, with log β5:1 = 22.5 ± 0.1, and show a very strong multivalent effect. The exTTFAuNP·C60 supramolecular associate represents a synthetically more accessible alternative to the large exTTF dendrimers we have reported previously.6c Such nanostructured donor–acceptor composites hold promise for their application in photovoltaic devices. The investigation of this possibility will be the subject of our future research.
Acknowledgements
We thank F. J. García de Abajo for generously providing the software for the simulation of the UV-vis spectra of the nanoparticles. Financial support by the MICINN of Spain (CTQ2008-00795/BQU, PIB2010JP-00196, and CSD2007-00010), FUNMOLS (FP7-212942-1), and the CAM (MADRISOLAR-2 S2009/PPQ-1533) is acknowledged. HI and EMP thank the MICINN for a FPU studentship and a Ramón y Cajal fellowship cofinanced by the European Social Fund, respectively.Notes and references
- For recent reviews, see: (a) E. M. Pérez, Pure Appl. Chem., 2011, 83, 201 CrossRef; (b) H. B. Liu, J. L. Xu, Y. J. Li and Y. L. Li, Acc. Chem. Res., 2010, 43, 1496 CrossRef CAS; (c) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (d) E. M. Pérez, B. M. Illescas, M. A. Herranz and N. Martín, New J. Chem., 2009, 33, 228 RSC; (e) L. Zang, Y. K. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS.
- For selected recent examples, see: (a) L. Welte, A. Calzolari, R. Di Felice, F. Zamora and J. Gomez-Herrero, Nat. Nanotechnol., 2010, 5, 110 CrossRef CAS; (b) X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Adrienko, K. Kremer and K. Müllen, Nat. Mater., 2009, 8, 421 CrossRef CAS; (c) J. M. Mativetsky, M. Kastler, R. C. Savage, D. Gentilini, M. Palma, W. Pisula, K. Müllen and P. Samori, Adv. Funct. Mater., 2009, 19, 2486 CrossRef CAS; (d) X. Feng, W. Pisula, T. Kudernac, D. Wu, L. Zhi, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2009, 131, 4439 CrossRef CAS; (e) F. D'Souza, E. Maligaspe, K. Ohkubo, M. E. Zandler, N. K. Subbaiyan and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 8787 CrossRef CAS; (f) G. Bottari, D. Olea, C. Gómez-Navarro, F. Zamora, J. Gómez-Herrero and T. Torres, Angew. Chem., Int. Ed., 2008, 47, 2026 CrossRef CAS.
- (a) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (b) M. M. S. Abdel-Mottaleb, E. Gomar-Nadal, M. Surin, H. Uji-i, W. Mamdouh, J. Veciana, V. Lemaur, C. Rovira, J. Cornil, R. Lazzaroni, D. B. Amabilino, S. De Feyter and F. C. De Schryver, J. Mater. Chem., 2005, 15, 4601 RSC; (c) J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS.
- (a) S. S. Gayathri, M. Wielopolski, E. M. Pérez, G. Fernández, L. Sánchez, R. Viruela, E. Ortí, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2009, 48, 815 CrossRef CAS; (b) E. M. Pérez, A. L. Capodilupo, G. Fernández, L. Sánchez, P. M. Viruela, R. Viruela, E. Ortí, M. Bietti and N. Martín, Chem. Commun., 2008, 4567 RSC; (c) E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc., 2006, 128, 7172 CrossRef CAS.
- (a) E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512 RSC; (b) D. Canevet, M. Gallego, H. Isla, A. de Juan, E. M. Pérez and N. Martín, J. Am. Chem. Soc., 2011, 133, 3184 CrossRef CAS; (c) B. Grimm, J. Santos, B. M. Illescas, A. Munoz, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2010, 132, 17387 CrossRef CAS; (d) E. Huerta, H. Isla, E. M. Pérez, C. Bo, N. Martín and J. de Mendoza, J. Am. Chem. Soc., 2010, 132, 5351 CrossRef CAS; (e) H. Isla, M. Gallego, E. M. Pérez, R. Viruela, E. Ortí and N. Martín, J. Am. Chem. Soc., 2010, 132, 1772 CrossRef CAS; (f) J. Santos, B. Grimm, B. M. Illescas, D. M. Guldi and N. Martín, Chem. Commun., 2008, 5993 RSC; (g) E. M. Pérez, M. Sierra, L. Sánchez, M. R. Torres, R. Viruela, P. M. Viruela, E. Ortí and N. Martín, Angew. Chem., Int. Ed., 2007, 46, 1847 CrossRef CAS.
- (a) G. Fernández, E. M. Pérez, L. Sánchez and N. Martín, Angew. Chem., Int. Ed., 2008, 47, 1094 CrossRef CAS; (b) G. Fernández, E. M. Pérez, L. Sánchez and N. Martín, J. Am. Chem. Soc., 2008, 130, 2410 CrossRef CAS; (c) G. Fernández, L. Sánchez, E. M. Pérez and N. Martín, J. Am. Chem. Soc., 2008, 130, 10674 CrossRef CAS; (d) J. Santos, E. M. Pérez, B. M. Illescas and N. Martín, Chem.–Asian J., 2011 DOI:10.1002/asia.201000832.
- For reviews, see: (a) U. H. F. Bunz and V. M. Rotello, Angew. Chem., Int. Ed., 2010, 49, 3268 CAS; (b) D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem. Int. Ed., 2010, 49, 3280 CAS. For recent examples, see: (c) R. Cao and B. Li, Chem. Commun., 2011, 47, 2865 RSC; (d) N. Kanayama, T. Takarada and M. Maeda, Chem. Commun., 2011, 47, 2077 RSC; (e) D. Feng, Y. Zhang, W. Shi, X. Li and H. Ma, Chem. Commun., 2010, 46, 9203 RSC; (f) Z. Zhu, C. Wu, H. Liu, Y. Zou, X. Zhang, H. Kang, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2010, 49, 1052 CrossRef CAS; (g) X. Xu, W. L. Daniel, W. Wei and C. A. Mirkin, Small, 2010, 6, 623 CrossRef CAS; (h) Y. Jiang, H. Zhao, Y. Lin, N. Zhu, Y. Ma and L. Mao, Angew. Chem., Int. Ed., 2010, 49, 4800 CAS; (i) X. Li, J. Wang, L. Sun and Z. Wang, Chem. Commun., 2010, 46, 988 RSC.
- (a) J. Ohyama, Y. Hitomi, Y. Higuchi, M. Shinagawa, H. Mukai, M. Kodera, K. Teramura, T. Shishido and T. Tanaka, Chem. Commun., 2008, 6300 RSC; (b) M. Kanehara, H. Takahashi and T. Teranishi, Angew. Chem., Int. Ed., 2008, 47, 307 CrossRef CAS; (c) T. Akiyama, M. Nakada, N. Terasaki and S. Yamada, Chem. Commun., 2006, 395 RSC; (d) H. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano and S. Isoda, Adv. Mater., 2005, 17, 1727 CrossRef CAS; (e) T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216 CrossRef CAS; (f) H. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, M. Isosomppi, N. V. Tkachenko and H. Lemmetyinen, Chem.–Eur. J., 2005, 11, 7265 CrossRef CAS.
- (a) G. Zhang, D. Zhang, X. Zhao, X. Ai, J. Zhang and D. Zhu, Chem.–Eur. J., 2006, 12, 1067 CrossRef CAS; (b) W.-J. Guo, J. Dai, D.-Q. Zhang, Q.-Y. Zhu and G.-Q. Bian, Inorg. Chem. Commun., 2005, 8, 994 CrossRef CAS; (c) J. Dai, L. Guo, Y. Jiang, Q.-Y. Zhu, R.-A. Gu, D.-x. Jia and W.-J. Guo, J. Nanosci. Nanotechnol., 2005, 5, 474 CrossRef CAS; (d) H. Nakai, M. Yoshihara and H. Fujihara, Langmuir, 1999, 15, 8574 CrossRef CAS.
- G. Frens, Nature, 1973, 241, 20 CAS.
- U. Kreibig and L. Genzel, Surf. Sci., 1985, 156, 678 CrossRef.
- For other examples of supramolecular systems based on the exTTF-C60 interaction, see: (a) X. Yang, G. Zhang, D. Zhang and D. Zhu, Langmuir, 2010, 26, 11720 CrossRef CAS; (b) X. Yang, G. Zhang, D. Zhang, J. Xiang, G. Yang and D. Zhu, Soft Matter, 2011, 7, 3592 RSC.
- We have followed the methodology described in: (a) F. J. García de Abajo and A. Howie, Phys. Rev. Lett., 1998, 80, 5180 CrossRef CAS; (b) F. J. García de Abajo and A. Howie, Phys. Rev. B: Condens. Matter, 2002, 65, 115418-1–17; (c) V. Myroshnychenko, J. Rodríguez-Fernández, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzán and F. J. García de Abajo, Chem. Soc. Rev., 2008, 37, 1792 RSC; (d) V. Myroshnychenko, E. Carbó-Argibay, I. Pastoriza-Santos, J. Pérez-Juste, L. M. Liz-Marzán and F. J. García de Abajo, Adv. Mater., 2008, 20, 4288 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- Minimizations were carried out utilizing the AMBER force field included in the HyperChem molecular modelling system. The AMBER force field was selected because it includes a term for van der Waals and electrostatic interactions. See: W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 Search PubMed.
- D. V. Konarev, R. N. Lyubovskaya, N. V. Drichko, E. I. Yudanova, Y. M. Shul'ga, A. L. Litvinov, V. N. Semkin and B. P. Tarasov, J. Mater. Chem., 2000, 10, 803 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and experimental details. See DOI: 10.1039/c1sc00179e |
| This journal is © The Royal Society of Chemistry 2011 |