Memory of chirality in J-type aggregates of an achiral perylene dianhydride dye created in a chiral asymmetric catalytic synthesis†
Jin-Song
Zhao
,
Yi-Bin
Ruan
,
Rong
Zhou
and
Yun-Bao
Jiang
*
Department of Chemistry, College of Chemistry and Chemical Engineering, and the MOE Key Laboratory of Analytical Sciences, Xiamen University, Xiamen, 361005, China. E-mail: ybjiang@xmu.edu.cn; Fax: +86 592 218 6401; Tel: +86 592-218 8372
First published on 25th February 2011
Abstract
Perylene dyes tend to form H-aggregates because of their large aromatic π-core and high hydrophobicity. We report here a chiral asymmetric catalytic synthesis of water-soluble chiral J-aggregates from highly hydrophobic and achiral perylene dianhydride (PDA). We propose to generate in situ aggregating hydrophobic species from a water soluble precursor so that it aggregates at a low concentration in solution within a predesigned microenvironment to stabilize the formed aggregates. Choosing PDA as an example, we generated it in situ from a water-soluble perylene-3,4,9,10-tetracarboxylate in acidic cetyltrimethylammonium bromide (CTAB) micelle solution, employing small molecule D- and/or L-tartaric acid as the chiral auxiliary. CD spectra of the PDA J-aggregates formed in the presence of D- and L-tartaric acid, respectively, were mirror images of each other. When the chiral auxiliary was absent, water-soluble J-aggregates of PDA were also obtained from acidic HCl-CTAB solution but they were then achiral. This confirms that the chirality of the J-aggregates is transferred from tartaric acid. Using an L-/D-tartaric acid mixture of varying ee as the chiral auxiliary, the exciton-coupled induced circular dichroism (ICD) signal of the formed PDA J-aggregates shows an “S”-shaped profile when plotted versus ee, indicating that chiral amplification occurs. Purified J-aggregates were found to contain PDA and CTAB, whereas tartaric acid was hardly detected, yet the ICD signals remained the same as those in the presence of tartaric acid before purification. This indicates memory in the J-aggregates of the chirality of tartaric acid. The ICD signal of the purified J-aggregates was also found to remain unchanged upon addition of a large excess of the other enantiomer of tartaric acid than the one used to induce the chirality, implying the imprinting of the chirality in the J-aggregates. The J-aggregates of PDA were shown to be positively charged from the polar heads of the CTAB surfactant molecules that help to disperse the aggregates and prevent their precipitation in aqueous solution. It was found that it is critical that the surfactant tail is long enough for the chiral J-aggregates to be created. This suggests that the hydrophobic tail might act as a pillar to support the J-aggregates that are driven by edge-edge hydrophobic interactions. With this in situ generation for the highly hydrophobic perylene dyes we succeeded in creating water soluble J-aggregates, chiral and achiral, in a controllable manner.
Introduction
Fabricating supramolecular structures with memory of chirality has drawn intensive recent interest because of its theoretical significance and potential applications.1–3 For memory of the chirality, chiral induction and memorization are two key processes. The former process transfers the chirality from chiral auxiliary to the supramolecular structure formed by achiral components, while the latter renders the supramolecular structure with the ability to sustain the chirality after removal of the external chiral auxiliary. Memory of chirality has been successfully achieved in macromolecules.4 Construction of supramolecular structures with memorized chirality, despite successful to some extent, still remains a challenge since supramolecular structure that is maintained by weak non-covalent interactions is highly susceptible to dissociation and racemization. Memory of chirality requires kinetically inert structure that to a high extent relies on structural rigidity.5Hydrogen bonding,1 ion pairing,2 and metal coordination3 are directional forces that have been employed to enhance the rigidity of the formed supramolecular structures. Other non-covalent interactions, such as directionless hydrophobic interactions that widely exist in Nature,6 are rarely employed in that regard. Yashima et al.7 have created chiral J-aggregates from achiral charged cyanine dyes inside the hydrophobic helical cavity of a poly(phenylacetylene) polymervia weak hydrophobic interactions, in which the memory of the chirality in the J-aggregates was attributed to van der Waals interactions between the charged cyanine dyes. Herein, we report the transferring, amplification, and memory of chirality in J-type aggregates of achiral perylene-3,4,9,10-tetracarboxylic dianhydride (PDA, Scheme 1), in which hydrophobic interactions lead to enhanced structural rigidity that helps to sustain the chirality from the small molecule chiral auxiliary. This may be of significance for the understanding of the role that weak and directionless hydrophobic interactions play in the memory of chirality. | ||
| Scheme 1 Structure of PDA, PTK, L-/D-tartaric acid, L-lactic acid, and CTAB. | ||
Fabrication of supramolecular structures is in general carried out in solution phase.8Self-assembly of neutral perylene dyes has so far been carried out almost entirely in organic solvents and synthetic introduction of long alkyl chains is essential to increase the solubility of the neutral perylene dyes. Although the synthetic procedures to obtain perylene derivatives are not simple, investigation of the aggregation of perylene dyes has attracted intense interest due to the outstanding optical properties and potential applications of their aggregates in n-type semiconductors,9 photo-induced electron transfer systems,10 organic light-emitting diodes (OLEDs),11 and organic solar cells.12 Differing from cyanine dyes7,13 and porphyrins14 that easily form J-type aggregates, neutral perylene dyes tend to undergo “face-to-face” H-aggregation because of their strong π–π stacking,15 construction of their “head-to-tail” J-type aggregates being hence rather difficult. Würthner et al.16 have fabricated a series of J-aggregates of perylene dyes in methylcyclohexanevia strongly slipped packing of the perylene bisimides with the perylene core being twisted by bay substitution. Yagai et al.17 constructed H- to J-type transformable aggregates of core-unsubstituted perylene bisimides by hydrogen-bonding directed complexation with cyanuric acids of the melamine unit in perylene bisimide in methylcyclohexane. In these systems, non-polar organic solvents are essential for increasing the solubility of the perylene dyes and for facilitating intermolecular hydrogen bonding interaction. J-type aggregation of the neutral perylene dyes has, however, not been observed in aqueous solutions. This could be due to the facts that (i) perylene dyes, especially the non-functionalized perylene dianhydride, is poorly soluble in polar solvents such as water, which inhibits further assembling to form supramolecular structures, and (ii) even when the aggregation can occur it proceeds too fast to be controlled.
Enlightened by the dehydration of perylene tetracarboxylic acid that leads to perylene anhydride, a reaction that has been widely employed in the synthesis of perylene bisimide dyes,9c,18 we proposed to generate in situPDA from a water soluble precursor potassium perylene tetracarboxylate (PTK, Scheme 1). Tartaric acid was employed as a chiral auxiliary and an acid source. The dehydration reaction of the protonated PTK in acidic solution is slow, hence allowing for the in situ generation of PDA monomers to form asymmetric supramolecular structures under the mediation of chiral tartaric acid. Because of the weak intermolecular hydrogen bonding between tartaric acid and PDA in aqueous solution, chiral tartaric acid could easily detach from the formed chiral aggregates of PDA and it thus functions as a chiral catalyst in the dehydration-aggregation process. Neither the neutral perylene tetracarboxylic acid nor the subsequently formed PDA are soluble in water, CTAB micelles (Scheme 1) were therefore introduced in an attempt to prevent quick precipitation of the rapidly aggregated neutral perylene dyes, in which H-aggregates are dominant. We thus successfully fabricated the water-soluble J-aggregates of PDA, with amplified supramolecular chirality being memorized and imprinted.
Results and discussion
Chirality transfer in J-aggregates of PDA
Upon mixing PTK (50 μM) and L- or D-tartaric acid (0.5 mM) in CTAB (5 mM, critical micelle concentration ca. 0.9 mM) aqueous solution (see experimental section in ESI†), the original bright green luminescent solution changed to orange and then to a transparent magenta color gradually within 36 h at 17 °C. During this process the solution changed to be hardly luminescent. Formation of PDA was confirmed by the electrospray ionization mass spectroscopy (Fig. S1 in ESI†).Perylene dyes tend to undergo sandwich-like H-type aggregation, characterized by a blue-shift and broadening of the absorption band.15 However, the absorption spectrum of the in situ generated aggregates of PDA in aqueous CTAB micellar solution exhibits an unusually narrow band at 538 nm with a high extinction coefficient of 4.9 × 104 M−1 cm−1 (Fig. 1c).19 Injection of the generated aggregates of PDA (50 μM) in CTAB solution into DMF resulted in a bright yellow luminescent and temporarily stable solution that emitted the fluorescence typical of the perylene dianhydride dye monomer (Fig. S2, ESI†). In contrast to the absorption of the PDA monomer in DMF (Fig. 1b), absorption of its aggregates in CTAB solution is red-shifted by 21 nm from that of the monomer peaked at 517 nm, with an extraordinarily reduced full-width-at-half-maximum (FWHM) of 569 cm−1 compared to 1169 cm−1 of the monomer. These spectral features are indicative of the J-aggregation of PDA in aqueous CTAB micelle solution.16,17,20
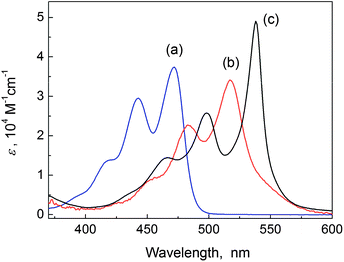 | ||
| Fig. 1 Absorption spectra of (a) PTK (50 μM) in aqueous CTAB (5 mM) solution, (b) PDA (2.5 μM) monomer in DMF, and (c) PDA (50 μM) J-aggregates in aqueous CTAB (5 mM) solution. | ||
A highly efficient chiral induction in the formed J-aggregates of PDA was identified from the appearance of strong split CD Cotton effects corresponding to the absorption of the J-aggregates mediated by chiral tartaric acid. The CD spectrum of J-aggregates mediated by L-tartaric acid (Fig. 2) exhibits two very strong Cotton effects of opposite signs in the absorption region around 538 nm of the J-band of PDA aggregates. The first Cotton effect at longer wavelength (543 nm) has a value Δε of +280.2 M−1 cm−1, while the second one at a shorter wavelength (530 nm) has a Δε value of −137.6 M−1 cm−1. These are typical features of exciton-coupled CD.21 Difference of the amplitudes of the two Cotton effects gives an A-value of 417.8 M−1cm−1.22 The positive A-value suggests a right-handed helical arrangement of the transition dipole moments of PDA molecules in the J-aggregates. The CD patterns of the J-aggregates of PDA mediated by L- and D-tartaric acids are perfect mirror images of each other (Fig. 2a), confirming that the induced CD signals of the J-aggregates of PDA originate from the absolute configuration of the chiral auxiliary tartaric acid.
![(a) CD spectra of PDA J-aggregates mediated by l- (black trace) and d-tartaric acid (red trace) and (b) absorption spectrum of PDA J-aggregates. Concentration of PDA was 50 μM in aqueous CTAB (5 mM) solution. [tartaric acid] = 0.5 mM.](/image/article/2011/SC/c1sc00043h/c1sc00043h-f2.gif) | ||
| Fig. 2 (a) CD spectra of PDA J-aggregates mediated by L- (black trace) and D-tartaric acid (red trace) and (b) absorption spectrum of PDA J-aggregates. Concentration of PDA was 50 μM in aqueous CTAB (5 mM) solution. [tartaric acid] = 0.5 mM. | ||
In order to follow the formation of the chiral J-aggregates, the dehydration reaction from PTK to the J-aggregates of PDA was monitored by both absorption and CD spectra (Fig. 3). At the time of mixing PTK with the chiral tartaric acid, when the generated PDA may have not aggregated, no CD signal was observed which suggests that the chirality can not be transferred from the chiral auxiliary to the perylene dye monomers. As the reaction proceeded, the original vibronic absorption of the monomers decreased while a new band at longer wavelength emerged (Fig. 3b). However, a CD signal was still not observed in the first 6 h. Only thereafter a distinct sharp absorption at 538 nm evolved as well as the exciton-coupled CD signal (Fig. 3a), indicating the start of the J-type chiral aggregation of the in situ generated PDA. It is noteworthy that during the aggregation process the shape of the CD spectrum remains the same (Fig. 3a). This means that it is the ultimately formed J-aggregates rather than any intermediate form that exhibit the induced chirality.
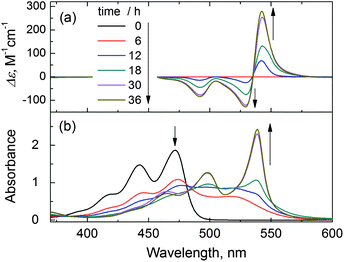 | ||
| Fig. 3 Traces of (a) CD and (b) absorption spectra during the J-aggregation process of PDA (50 μM) (mediated by L-tartaric acid, 0.5 mM) in aqueous CTAB (5 mM) solution at 17 °C within 36 h. | ||
Chiral amplification in the in situ created J-aggregates
Chiral amplification in supramolecular structures has inspired a great variety of research because of its significance in understanding the origin of chirality in Nature and the applications in asymmetric synthesis.23 We examined the induced supramolecular chirality of the J-aggregates as a function of the ee of the chiral auxiliary of L- and D-tartaric acids mixture. The induced CD signal of the J-aggregates of PDA was found to exhibit an “S”-shaped profile when plotted against the ee (Fig. 4). This means a major enantiomer of the chiral auxiliary dictates the helical handedness of the supramolecular assembly, known as the “majority-rules”.23,24 For example, L-tartaric acid at only 20% ee is able to induce a CD signal 58% as large as that observed with pure L-tartaric acid (Fig. 4). A chiral amplification in the PDA J-aggregates is hence shown.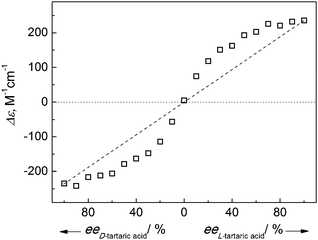 | ||
| Fig. 4 Δε value at 543 nm of PDA (50 μM) J-aggregates mediated by tartaric acid (0.5 mM) of varying ee value in aqueous CTAB (5 mM) solution. Dashed line indicates the response in case of no chiral amplification. | ||
Role of tartaric acid in chiral induction in the J-aggregates
These observations suggested that a chiral induction occurred by tartaric acid to the J-aggregates of neutral PDA upon the in situ generation of PDA. The influence of tartaric acid concentration on the chiral induction was investigated. In order to fix the extent of PTK protonation, the solution pH was maintained at 3.0 by HCl or KOH. Varying amounts of tartaric acid were introduced, from 0 to 100 equivalents of PTK. In the absence of tartaric acid, the aggregates formed in the acidic solution of HCl (pH 3.0) showed the same absorption spectra as those of the J-aggregates formed in the acidic solutions of chiral tartaric acids, whereas no induced CD signal was observed. This observation indicated that PDA generated in situ in acidic CTAB solutions from water-soluble PTK could form J-aggregates, irrespective of the presence or not of the chiral auxiliary. The fact that when chiral tartaric acid was added, the obtained J-aggregates of PDA were chiral, confirmed that the chirality was transferred from the tartaric acid. In this case, the induced supramolecular chirality of the J-aggregates is highly dependent on the concentration of tartaric acid. With increasing concentration of tartaric acid from 2 to 10 equivalents of PTK, the intensity of the induced CD signal was enhanced by over 17-fold from 16.4 to 286.7 M−1 cm−1 (Fig. 5), although the absorption spectra are more or less the same. The nonlinear profile of the induced CD signal towards the concentration of chiral auxiliary in an “S”-shape (Fig. 5 inset), together with the observed “majority-rules” character shown in Fig. 4, suggests a cooperative interaction in the chiral inducing process. Further increase in the concentration of tartaric acid (>20 equivalents) disfavored the formation of J-aggregates, both the J-band at 538 nm and the corresponding CD signal decreased (Fig. 5 inset).![CD spectra of PDA J-aggregates mediated by l-tartaric acid of varying concentration in pH 3.0 aqueous CTAB solution (5 mM). Inset shows the CD intensity at 543 nm as a function of [l-tartaric acid]/[PTK] ratio. [PDA] = 50 μM.](/image/article/2011/SC/c1sc00043h/c1sc00043h-f5.gif) | ||
| Fig. 5 CD spectra of PDA J-aggregates mediated by L-tartaric acid of varying concentration in pH 3.0 aqueous CTAB solution (5 mM). Inset shows the CD intensity at 543 nm as a function of [L-tartaric acid]/[PTK] ratio. [PDA] = 50 μM. | ||
Control experiments with a chiral model acid, L-lactic acid that bears only one carboxylic group and one chiral carbon (Scheme 1), showed that only an insignificant induced CD signal was observed, the intensity being less than 3% of that induced by L-tartaric acid, while the absorption spectra were the same as those of the J-aggregates mediated by the chiral tartaric acids (Fig. S3, ESI†). This observation strongly supports the critical role of the two asymmetrically arranged carboxylic groups of chiral tartaric acid in inducing the supramolecular chirality in the PDA J-aggregates. Referring to the structure of the tartaric acid (Scheme 1), rotation of the two chiral centers around the C*–C* axis leads to three conformers, T, G+, and G− (Fig. 6).25 According to the exciton chirality model,21 the two carboxylic groups arranged in one plane in the preferred T conformer of the tartaric acid would not be able to induce any exciton-coupled CD of PDA. The G+ conformer of L-tartaric acid, despite a higher steric hindrance from the adjacent carboxylic groups, could be stabilized by the two possible six-membered ring intramolecular hydrogen bonds.25a An equilibrium therefore exists between the T and G+ conformers of tartaric acid. Arrangement of PDA monomers along the direction of the two carboxylic groups in the G+ conformer of L-tartaric acid would lead to a right-handed helical sense that will induce a positive exciton-coupled CD of the PDA J-aggregates (Fig. 6),21 which is indeed confirmed by the experimental observation (Fig. 2a, black trace).
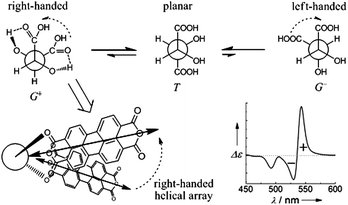 | ||
| Fig. 6 Conformation equilibrium of L-tartaric acid and the induced right-handed helical array of PDA due to the G+ conformer of L-tartaric acid. The right-handed helical arrangement of the transition dipole moments of PDA monomers would induce a positive Cotton effect at longer wavelength and a negative Cotton effect at shorter wavelength, known as the positive exciton-coupled CD illustrated in the CD spectrum. | ||
Memory of the induced supramolecular chirality in J-aggregates of PDA
It was observed that once the aggregate was formed, the chirality was “memorized”. The positive exciton-coupled CD signal of the PDA J-aggregates induced by L-tartaric acid remained unchanged after further addition of 10-fold excess of D-tartaric acid, even after later standing at 10–15 °C for 1 week when over 95% of its original CD intensity was observed (Fig. S4, see ESI†). Meanwhile, achiral PDA J-aggregates formed in HCl-CTAB solution remained achiral upon later addition of excess amounts of chiral tartaric acid. These observations show that the chirality of the formed J-aggregates of PDA is stable enough to resist the disturbance of external chiral stimulus.The J-aggregates of PDA were further isolated and purified by means of precipitation (see experimental section in ESI†). The purified J-aggregates exhibit very broad 1H NMR signals in D2O (Fig. S5a, see ESI†), suggesting a compact aggregation of the components. Upon addition of excess amounts of solid KOH, hydrolysis of PDA to PTK occurred immediately and the 1H NMR spectrum then showed distinct PTK and CTAB proton signals (Fig. S5b, see ESI†) whereas no C*–H signal of tartrate at 4.23 ppm was observed (Fig. S6, see ESI†). The J-aggregates were then probed to constitute of PDA and CTAB. On the basis of the integrals of proton peaks the molar ratio of PDA to CTAB was calculated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 (Fig. S5b, see ESI†). 1H NMR titrations of L-tartrate in D2O solution of PTK showed that the amount of the possibly remaining tartaric acid was less than 0.5% of PDA (Fig. S7, see ESI†), that should have not been enough to induce the chirality of the aggregates if the tartaric acid was indeed needed (Fig. 5). Actually we found that the absorption and CD spectra of the purified J-aggregates were almost the same as those before purification when excess amounts of tartaric acid and CTAB coexisted (Fig. S8, see ESI†). This means that the chirality of the J-aggregates of PDA is memorized and imprinted. Further addition of D-tartaric acid in the purified chiral J-aggregates of PDA mediated by L-tartaric acid led to no changes in the CD spectrum (Fig. S9, see ESI†), further confirming the chirality imprinting in the PDA J-aggregates. These observations actually reveal the catalytic nature of the tartaric acid in chiral induction, since the tartaric acid is neither a part of the finally synthesized chiral product (the aggregates), nor being changed during the formation of the aggregates. Refering to the previously reported examples of memory of chirality in supramolecular aggregates from achiral species,1–3,7 such a chiral asymmetric synthetic approach for chiral J-aggregates of perylene dyes, together with the chiral amplification and memory, represents the first of its kind.
:1.6 (Fig. S5b, see ESI†). 1H NMR titrations of L-tartrate in D2O solution of PTK showed that the amount of the possibly remaining tartaric acid was less than 0.5% of PDA (Fig. S7, see ESI†), that should have not been enough to induce the chirality of the aggregates if the tartaric acid was indeed needed (Fig. 5). Actually we found that the absorption and CD spectra of the purified J-aggregates were almost the same as those before purification when excess amounts of tartaric acid and CTAB coexisted (Fig. S8, see ESI†). This means that the chirality of the J-aggregates of PDA is memorized and imprinted. Further addition of D-tartaric acid in the purified chiral J-aggregates of PDA mediated by L-tartaric acid led to no changes in the CD spectrum (Fig. S9, see ESI†), further confirming the chirality imprinting in the PDA J-aggregates. These observations actually reveal the catalytic nature of the tartaric acid in chiral induction, since the tartaric acid is neither a part of the finally synthesized chiral product (the aggregates), nor being changed during the formation of the aggregates. Refering to the previously reported examples of memory of chirality in supramolecular aggregates from achiral species,1–3,7 such a chiral asymmetric synthetic approach for chiral J-aggregates of perylene dyes, together with the chiral amplification and memory, represents the first of its kind.
The purified PDA-CTAB binary aggregates were found to be stable in water at 10–15 °C for several weeks, the CD signal at 543 nm being 70% of the original value after one month. However, the J-aggregates became unstable at a higher temperature of 30 °C, when a transformation of the J-aggregates to H-aggregates occurred. This was indicated by the time-dependent absorption spectrum. The J-band at 538 nm of the J-aggregates was found to gradually disappear while a new and substantially blue-shifted and broad band at 469 nm (the H-band) developed (Fig. 7b). Meanwhile, the CD signals drastically decreased and eventually inverted to a weak and negative exciton-coupled CD signal (Fig. 7a).
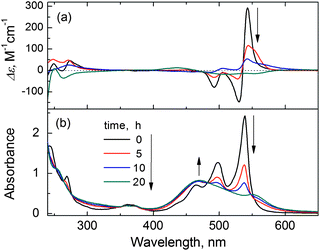 | ||
| Fig. 7 Traces of (a) CD and (b) absorption spectra of the purified PDA (50 μM) J-aggregates (mediated by L-tartaric acid) in water at 30 °C within 20 h. | ||
X-ray diffraction (XRD) of the J-aggregates showed no significant diffraction signal, whereas the transformed H-aggregates exhibited distinct π–π stacking peak with a d-spacing of 3.24 Å (Fig. S10, see ESI†). The latter is consistent with the “face-to-face” character of the H-aggregation.9c The absence of a diffraction signal from the J-aggregates suggests no or little π–π stacking, which is interesting in terms of the strong π–π stacking nature of the large perylene core in PDA. However, PDA could alternatively take an “edge-to-edge” array in a coplanar pattern (Fig. 6 and Fig. 8 in the next section), leading to J-aggregates that structurally resemble helical organic nanotubes formed by coplanarly arranged naphthalenediimides bearing chiral amino acid residues.26 In the J-aggregates of PDA, perylene dyes may form similar tube-like structure maintained by hydrophobic interaction, while the tail of CTAB molecule could be stuffed in the tubular cavity acting like a pillar (Fig. 8 in the next section). This proposed pattern of aggregation could explain the broad 1H NMR bands (Fig. S5, see ESI†) and XRD data (Fig. S10, see ESI†) and account for the observed smaller spectral red-shift by 21 nm of the J-aggregates of PDA (Fig. 1) compared to a large red-shift (>70 nm) in the previously reported J-aggregates in organic solvents that are formed by π–π stacked perylene bisimide dyes in a slipped arrangement.16
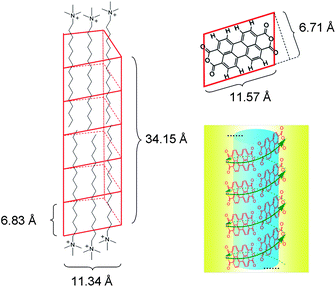 | ||
| Fig. 8 Modelling of the proposed structure of CTAB embedded PDA aggregates using Chem3D. Calculations show that the volume of the cavity formed by 15 PDA molecules (5 circles, 3 PDA molecules per circle) is 1912 Å3 that could accommodate ca. 26 CTAB alkyl chains, giving a 1:1.7 molar ratio of PDA to CTAB. The blue shadow represents the hydrophobic microenvironment in which CTAB is embedded and the yellow shadow represents the surrounding aqueous medium. | ||
Role of CTAB in the formation of chiral J-aggregates of PDA
A high concentration of CTAB (5 mM, cmc of CTAB is 0.9 mM) was originally applied to prevent the rapid aggregation of the highly hydrophobic neutral perylene species. At lower CTAB concentration, H-type aggregation of PDA was indeed observed that eventually led to precipitation. Detailed investigations revealed that the role of CTAB was beyond that originally intended. 1H NMR of the purified J-aggregates showed that CTAB was a component in the aggregates. The observed very broad NMR signals of CTAB between 0 and 4 ppm (Fig. S5a, see ESI†) were indicative of the compact aggregation of CTAB within a confined space, the tail of CTAB being something like a pillar to support the asymmetrically aligned PDA molecules in the J-aggregates. The zeta potential of the purified PDA J-aggregates in water, measured to be +36.72 mV, indeed informed that the aggregates were highly positively charged. This served as a support of the pillar role of CTAB in stabilizing the J-aggregates of PDA. The positive charges of the aggregates brought about by the included CTAB molecules thereby facilitated stable dispersion of the aggregates in water and prevented them from further aggregation. A structural rigidity was hence afforded as the adjacent PDA molecules were interlocked under the immense pressure of the hydrophobic effect and backed up by the embedded CTAB (Fig. 8), thereby diminishing racemization and rendering the J-aggregates with excellent memory of the induced chirality. The contribution of the hydrophobic effect was supported by the effect of the added organic solvents such as ethanol and acetonitrile into the aqueous solutions of the J-aggregates that led to a quick J- to H-aggregate transformation and a concomitant dramatic decline of the CD signals.Modeling of the CTAB embedded structure of the J-aggregates of PDA suggested that the aggregate composed of 15 PDA monomers (5 circles, based on CTAB tail-to-tail embedding pattern) was able to accommodate ca. 26 CTAB molecules (Fig. 8), giving a 1:1.7 molar ratio of PDA to CTAB. This may help to define the structure of the PDA J-aggregates we experimentally created, for which 1H NMR suggested a 1:1.6 molar ratio of PDA to CTAB (Fig. S5b, see ESI†).
In order to understand the “pillar”-like role of the alkyl tail in the surfactant molecule in backing up the J-aggregates of PDA, CTAB derivatives with tail of varying length were examined. The absorption and CD spectra of the PDA aggregates formed in the solution of 3 mM OcTAB with a C18 tail (cmc of OcTAB, 0.2 mM) are similar to those of the aggregates obtained in the solution of CTAB bearing a C16 tail (Fig. 9), respectively. The absorption spectra of the PDA aggregates obtained in the solution of 5 mM TTAB with a shorter C14 tail (cmc of TTAB, 3.8 mM), however, exhibited two distinct bands, the red-shifted one at 549 nm (the J-band) and the blue-shifted and broad band at 478 nm (the H-band) (Fig. 9b). This suggested that both the J- and H-type aggregation occurred in the solution of TTAB. The CD spectrum, although it exhibited a positive exciton-coupled CD signal corresponding to the J-band at 549 nm (Fig. 9a), gave an A-value of +98.3 M−1 cm−1 that is much lower than that of the aggregates obtained in CTAB solution (+417.8 M−1 cm−1, Fig. 2a). In the solution of DTAB (20 mM, cmc of DTAB 14.4 mM) with a further shorter C12 tail, the obtained PDA aggregates were dominantly of H-type, as suggested by a distinct blue-shifted and broad H-band absorption around 470 nm and a very weak CD signal (Fig. 9). Since the cmc values of these surfactants differ quite much, experiments at a variety of TTAB concentrations were then performed. It was found that with increasing TTAB concentration from 5 mM to 100 mM, the absorption at 513 nm of the PDA monomer was restored together with a total quenching of the CD signal (Fig. S11, see ESI†). This means that an increase in the concentration of the surfactant bearing a short tail does not help the J-aggregation, but instead inhibits the aggregation. It was hence shown that the alkyl tail of the surfactant molecule should not be shorter than C16 in order to afford the J-aggregates of PDA in the aqueous micellar solution, a fact that supports the pillar role of the surfactant tail.
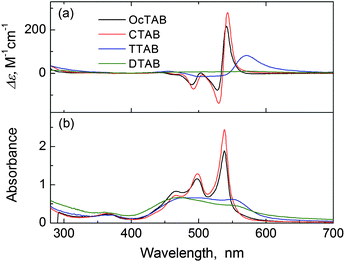 | ||
| Fig. 9 (a) CD and (b) absorption spectra of PDA (50 μM) aggregates mediated by L-tartaric acid (0.5 mM) in OcTAB (3 mM, black trace), CTAB (5 mM, red trace), TTAB (5 mM, blue trace), and DTAB (20 mM, green trace) solution. | ||
Conclusions
We developed a convenient and efficient chiral catalytic procedure for fabricating J-type aggregates from the highly hydrophobic perylene dianhydride in aqueous solution. A slow dehydration reaction that allows for an in situ generation of PDA from its water soluble precursor perylene tetracarboxylate in acidic CTAB solution was established to afford stable and water soluble J-aggregates in aqueous solutions. These were shown to be positively charged, led by the inclusion of cationic surfactant molecules in the aggregates. In agreement with this observation was the fact that stable J-aggregates could also be formed in the acidic solutions of OcTAB with a longer alkyl tail; whereas in the acidic solutions of TTAB and DTAB bearing shorter tails than C16, a mixture of J- and H-aggregates and only H-aggregates were obtained, respectively. This indicates that an alkyl tail in the surfactant molecule of sufficient length is necessary to support the J-aggregates of PDA. The CD spectra of the J-aggregates obtained in the solutions of L- and D-tartaric acids were mirror images of each other, confirming that the chirality of the J-aggregates was transferred from the tartaric acid. Using another chiral acid, L-lactic acid that bears only one carboxylic group, only a very weak CD signal was observed from the obtained J-aggregates of PDA, illustrating that both the two carboxylic groups of tartaric acid are involved in the asymmetric supramolecular aggregation of PDA for transferring the chirality of tartaric acid. The chirality of the obtained J-aggregates of PDA was found to be memorized and imprinted from the chiral auxiliary, as shown by the following facts, (i) the CD signals of the PDA J-aggregates remained unchanged after later addition of excess amounts of the chiral tartaric acid in the solutions where the aggregates were created in the presence of the tartaric acid of opposite chirality, (ii) the CD signals of the purified J-aggregates, probed to constitute of PDA and CTAB, remained to be the same as those before purification when tartaric acid co-existed, and (iii) later addition of excess amounts of tartaric acid of the opposite chirality to the purified J-aggregates of PDA did not lead to any change in the CD signals either. It is interesting that, when the acid source in the acidic CTAB solution of PTK was HCl, water soluble J-aggregates of PDA were obtained too. But the thus obtained J-aggregates from achiral PDA remained achiral, even after later addition of chiral tartaric acid. Again this supports that the chirality of the obtained J-aggregates of PDA in the presence of tartaric acid was derived from the chiral catalysis of the tartaric acid in the in situ generation and the chiral aggregation of PDA. Using a D- and L-tartaric acid mixture as the chiral auxiliary, the CD signal of the obtained chiral J-aggregates of PDA exhibited an “S”-shaped profile when plotted against the ee of the mixture, a phenomenon termed “majority-rule” that probes a chiral amplification in the chirality transferring. This is of significance for asymmetric organic synthesis and is probably of relevance to the understanding of the origin of chirality in Nature. Cationic surfactants with a sufficiently long alkyl tail were shown to play an important role in the slow creation of the highly hydrophobic PDA in aqueous solution. They also help to stabilize the formed J-aggregates, in which they are included, so that the alkyl chain acts like a pillar and its cationic polar head helps to disperse the otherwise hydrophobic aggregates in aqueous solutions. The present observation of chirality transfer, memory and imprinting, and amplification in the same system represents the first example that all these three phenomena are observed in aqueous solution of J-aggregates driven by directionless hydrophobic interactions. Given the importance of the aggregates of perylene derivatives and their applications in optoelectronics, chiral J-aggregates that are otherwise not feasible to create might be of significance for chiroptics and chiral optoelectronics. It is in this regard expected that the protocol of creating water soluble J-aggregates from highly hydrophobic perylene dyes in micellar solutions might be applied for other derivatives of perylenes.Acknowledgements
This work has been supported by the National Natural Science Foundation of China (Grant No. 20835005) and by the MOST of China (Grant No. 2011CB910400). We thank Prof. Hui Zhang of the Department of Chemistry, Xiamen University for her support in CD experiments.Notes and references
- (a) Y. Furusho, T. Kimura, Y. Mizuno and T. Aida, J. Am. Chem. Soc., 1997, 119, 5267–5268 CrossRef CAS; (b) Y. Mizuno, T. Aida and K. Yamaguchi, J. Am. Chem. Soc., 2000, 122, 5278–5285 CrossRef CAS; (c) Y. Mizuno and T. Aida, Chem. Commun., 2003, 20–21 RSC; (d) K. Toyofuku, M. A. Alam, A. Tsuda, N. Fujita, S. Sakamoto, K. Yamaguchi and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 6476–6480 CrossRef CAS; (e) J. M. Rivera, S. L. Craig, T. Martin and J. Rebek, Jr, Angew. Chem., Int. Ed., 2000, 39, 2130–2132 CrossRef CAS; (f) L. J. Prins, F. D. Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181–184 CrossRef CAS; (g) T. Ishi-i, M. Crego-Calama, P. Timmerman, D. N. Reinhoudt and S. Shinkai, J. Am. Chem. Soc., 2002, 124, 14631–14641 CrossRef CAS; (h) S. Váquez-Campos, M. Crego-Calama and D. N. Reinhoudt, Supramol. Chem., 2007, 19, 95–106 CrossRef CAS; (i) F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753–16755 CrossRef CAS.
- (a) E. Bellacchio, R. Lauceri, S. Gurrieri, L. M. Scolaro, A. Romeo and R. Purrello, J. Am. Chem. Soc., 1998, 120, 12353–12354 CrossRef CAS; (b) R. Lauceri, A. Raudino, L. M. Scolaro, N. Micali and R. Purrello, J. Am. Chem. Soc., 2002, 124, 894–895 CrossRef CAS; (c) A. Mammana, A. D'Urso, R. Lauceri and R. Purrello, J. Am. Chem. Soc., 2007, 129, 8062–8063 CrossRef CAS; (d) R. Randazzo, A. Mammana, A. D'Urso, R. Lauceri and R. Purrello, Angew. Chem., Int. Ed., 2008, 47, 9879–9882 CrossRef CAS; (e) H. Onouchi, T. Miyagawa, K. Morino and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 2381–2384 CrossRef CAS; (f) L. X. Zeng, Y. J. He, Z. F. Dai, J. Wang, Q. Cao and Y. L. Zhang, ChemPhysChem, 2009, 10, 954–962 CrossRef CAS; (g) J. A. Wang, D. D. Ding, L. X. Zeng, Q. A. Cao, Y. J. He and H. Zhang, New J. Chem., 2010, 34, 1394–1400 RSC.
- (a) A. Sugasaki, M. Ikeda, M. Takeuchi, A. Robertson and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1999, 3259–3264 RSC; (b) Y. Kubo, T. Ohno, J. Yamanaka, S. Tokita, T. Iida and Y. Ishimaru, J. Am. Chem. Soc., 2001, 123, 12700–12701 CrossRef CAS; (c) M. Ziegler, A. V. Davis, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 2003, 42, 665–668 CrossRef CAS; (d) N. C. Fletcher, C. Martin and H. J. Abraham, New J. Chem., 2007, 31, 1407–1411 RSC.
- (a) Z. N. Yu, X. H. Wan, H. L. Zhang, X. F. Chen and Q. F. Zhou, Chem. Commun., 2003, 974–975 RSC; (b) K. Maeda and E. Yashima, Top. Curr. Chem., 2006, 265, 47–88 CAS; (c) T. Kawauchi, J. Kumaki, A. Kitaura, K. Okoshi, H. Kusanagi, K. Kobayashi, T. Sugai, H. Shinohara and E. Yashima, Angew. Chem., Int. Ed., 2008, 47, 515–519 CrossRef CAS; (d) E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef CAS; (e) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS; (f) Y. Hase, K. Nagai, H. Iida, K. Maeda, N. Ochi, K. Sawabe, K. Sakajiri, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2009, 131, 10719–10732 CrossRef CAS; (g) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS; (h) E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2010, 3, 68–73.
- (a) R. Purrello, Nat. Mater., 2003, 2, 216–217 CrossRef CAS; (b) L. Rosaria, A. D'Urso, A. Mammana and R. Purrello, Chirality, 2008, 20, 411–419 CrossRef CAS; (c) A. Scarso and J. Rebek, Jr, Top. Curr. Chem., 2006, 265, 1–46 CAS.
- (a) A. Ben-Naim, Hydrophobic Interactions, Plenum Press, New York, 1980 Search PubMed; (b) D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS.
- T. Miyagawa, M. Yamamoto, R. Muraki, H. Onouchi and E. Yashima, J. Am. Chem. Soc., 2007, 129, 3676–3682 CrossRef CAS.
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS; (b) A. Ajayaghosh, S. J. George and A. P. H. J. Schenning, Top. Curr. Chem., 2005, 258, 83–118 CAS; (c) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS; (d) Z. J. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC; (e) C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC.
- (a) C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudholter, J. Am. Chem. Soc., 2000, 122, 11057–11066 CrossRef; (b) P. R. L. Malenfant, C. D. Dimitrakopoulos, J. D. Gelorme, L. L. Kosbar, T. O. Graham, A. Curioni and W. Andreoni, Appl. Phys. Lett., 2002, 80, 2517–2519 CrossRef CAS; (c) Y. Che, A. Datar, K. Balakrishnan and L. Zang, J. Am. Chem. Soc., 2007, 129, 7234–7235 CrossRef CAS; (d) V. Marcon, D. W. Breiby, W. Pisula, J. Dahl, J. Kirkpatrick, S. Patwardhan, F. Grozema and D. Andrienko, J. Am. Chem. Soc., 2009, 131, 11426–11432 CrossRef CAS.
- (a) E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen, F. Würthner and R. A. J. Jansses, J. Phys. Chem. A, 2004, 108, 6933–6937 CrossRef CAS; (b) A. Prodi, C. Chiorboli, F. Scandola, E. Iengo, E. Alessio, R. Dobrawa and F. Würthner, J. Am. Chem. Soc., 2005, 127, 1454–1462 CrossRef CAS; (c) M. S. Rodriguez-Morgade, T. Torres, C. Atienza-Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145–15154 CrossRef CAS; (d) Y. K. Che, X. M. Yang, G. L. Liu, C. Yu, H. W. Ji, J. M. Zuo, J. C. Zhao and L. Zang, J. Am. Chem. Soc., 2010, 132, 5743–5750 CrossRef CAS.
- (a) K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 1229–1233 CrossRef CAS; (b) A. K. Pandey and J. M. Nunzi, Appl. Phys. Lett., 2007, 90(26), 263508 CrossRef; (c) F. Jaiser, D. Neher, A. Meisel, H. G. Nothofer, T. Miteva, A. Herrmann, K. Müllen and U. Scherf, J. Chem. Phys., 2008, 129(11), 114901 CrossRef.
- (a) J. J. Dittmer, E. A. Marseglia and R. H. Friend, Adv. Mater., 2000, 12, 1270–1274 CrossRef CAS; (b) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS; (c) L. Tan, M. D. Curtis and A. H. Francis, Chem. Mater., 2004, 16, 2134–2141 CrossRef CAS; (d) M. Sommer and M. Thelakkat, Eur. Phys. J.: Appl. Phys., 2007, 36, 245–249.
- For examples of J-aggregates of cyanine dyes, see (a) U. DeRossi, S. Dähne, S. C. J. Meskers and H. P. J. M. Dekkers, Angew. Chem., Int. Ed. Engl., 1996, 35, 760–763 CAS; (b) T. D. Slavnova, H. Görner and A. K. Chibisov, J. Phys. Chem. B, 2007, 111, 10023–10031 CrossRef CAS; (c) Y. Z. Zhang, J. F. Xiang, Y. L. Tang, G. Z. Xu and W. P. Yan, ChemPhysChem, 2007, 8, 224–226 CrossRef CAS; (d) L. X. Zeng, Y. J. He, Z. F. Dai, J. Wang, C. Q. Wang and Y. G. Yang, Sci. China, Ser. B: Chem., 2009, 52, 1227–1234 CrossRef CAS, For chiral induction in other aggregates of cyanine dyes, see (e) H. Gorner, T. D. Slavnova and A. K. Chibisov, J. Phys. Chem. B, 2010, 114, 9330–9337 CrossRef CAS; (f) J. L. Seifert, R. E. Connor, S. A. Kushon, M. Wang and B. A. Armitage, J. Am. Chem. Soc., 1999, 121, 2987–2995 CrossRef CAS; (g) S. Sforza, E. Scaravelli, R. Corradini and R. Marchelli, Chirality, 2005, 17, 515–521 CrossRef CAS.
- (a) S. G. Jiang, L. Zhang and M. H. Liu, Chem. Commun., 2009, 6252–6254 RSC; (b) Y. F. Qiu, P. L. Chen and M. H. Liu, J. Am. Chem. Soc., 2010, 132, 9644–9652 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- For J-aggregates from PBIs with bay substitution: (a) T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS; (b) H. Wang, T. E. Kaiser, S. Uemura and F. Würthner, Chem. Commun., 2008, 1181–1183 RSC; (c) T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719–6732 CrossRef CAS, For J-aggregates from core-unsubstituted PBIs: (d) F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef; (e) S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343–11357 CrossRef CAS.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS.
- X. Yang, X. H. Xu and H. F. Ji, J. Phys. Chem. B, 2008, 112, 7196–7202 CrossRef CAS.
- The ε and Δε for the aggregates are given as the values per aggregate-bound monomer unit.
- (a) E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS; (b) G. Scheibe, Angew. Chem., 1936, 49, 563 Search PubMed; (c) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS; (d) H. Kuhn, C. Kuhn in J-Aggregates (Ed.: T. Kobayashi), World Scientific, Singapore, 1996, pp. 1–40 Search PubMed; (e) O.K. Kim, J. Je, G. Jernigan, L. Buckley and D. Whitten, J. Am. Chem. Soc., 2006, 128, 510–516 CrossRef CAS; (f) S. Gadde, E. K. Batchelor, J. P. Weiss, Y. H. Ling and A. E. Kaifer, J. Am. Chem. Soc., 2008, 130, 17114–17119 CrossRef CAS; (g) J. M. W. Chan, J. R. Tischler, S. E. Kooi, V. Bulovic and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 5659–5666 CrossRef CAS.
- (a) N. Harada, K. Nakanishi, Circular Dichroic Spectroscopy-Exciton Coupling in Organic Stereochemistry, University Science Books, Mill Valley, 1983 Search PubMed; (b) K. Nakanishi, N. Berova, R. Woody, Circular Dichroism: Principles and Applications, 2nd ed., Wiley-VCH, New York, 2000, pp. 337–382 Search PubMed.
- The A-value of exciton-coupled CD Cotten effect is defined as Δε1 – Δε2 where Δε1 and Δε2 are intensities of the first and second Cotton effects, respectively.
- (a) A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS; (b) V. K. Praveen, S. S. Babu, C. Vijayakumar, R. Varghese and A. Ajayaghosh, Bull. Chem. Soc. Jpn., 2008, 81, 1196–1211 CrossRef CAS.
- (a) J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS; (b) J. Van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef CAS; (c) W. Jin, T. Fukushima, M. Niki, A. Kosaka, N. Ishii and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10801–10806 CrossRef CAS; (d) A. Lohr and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 1232–1236 CrossRef CAS; (e) M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 611–619 CrossRef CAS; (f) M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 620–626 CrossRef CAS.
- (a) J. Gawroński, K. Gawrońska, P. Skowronek, U. Rychlewska, B. Warzajtis, J. Rychlewski, M. Hoffmann and A. Szarecka, Tetrahedron, 1997, 53, 6113–6144 CrossRef CAS; (b) R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS; (c) D. Berthier, T. Buffeteau, J. M. Léger, R. Oda and I. Huc, J. Am. Chem. Soc., 2002, 124, 13486–13494 CrossRef CAS; (d) B. V. Shankar and A. Patnaik, J. Phys. Chem. B, 2007, 111, 9294–9300 CrossRef CAS.
- (a) G. D. Pantoş, P. Pengo and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194–197 CrossRef CAS; (b) G. D. Pantoş, J. L. Wietor and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 2238–2240 CrossRef CAS; (c) B. M. Bulheller, G. D. Pantoş, J. K. M. Sanders and J. D. Hirst, Phys. Chem. Chem. Phys., 2009, 11, 6060–6065 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, ESI-MS, spectroscopic data and 1H NMR spectra. See DOI: 10.1039/c1sc00043h |
| This journal is © The Royal Society of Chemistry 2011 |