Development and evaluation of new cyclooctynes for cell surface glycan imaging in cancer cells†
Henning
Stöckmann
ab,
André A.
Neves
b,
Shaun
Stairs
a,
Heather
Ireland-Zecchini
b,
Kevin M.
Brindle
b and
Finian J.
Leeper
*a
aUniversity of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: fjl1@cam.ac.uk; Fax: +44 (0) 1223 336 362; Tel: +44 (0) 1223 336 403
bCancer Research UK Cambridge Research Institute, Li Ka Shing Centre, Robinson Way, Cambridge, CB2 0RE, UK
First published on 25th February 2011
Abstract
Two reagents have been synthesized for selective labeling of cell surface azidoglycans, an unusually stable version of a dibenzo cyclooctyne (TMDIBO) and a third-generation difluorinated cyclooctyne (DIFO3). Both syntheses are efficient with minimal purification, and the dibenzo cyclooctyne is stable under basic and acidic conditions. Flow cytometric measurements with azidosugar labeled cancer cells, in which these reagents were linked to the fluorophore Alexa Fluor 647, gave a signal-to-background ratio of up to 35 with TMDIBO as compared to ≈10 for DIFO3 and ≈5 for a phosphine reagent. TMDIBO-based probes should have applications in molecular imaging of cell surface glycans in vivo.
Introduction
Altered glycosylation patterns of cell surface proteins are a hallmark of the tumour phenotype. Bertozzi and co-workers have pioneered a novel approach to glycan imaging, where cells, or even whole animals, are exposed to peracetylated azido-labeled sugars and these get incorporated into cell surface glycans.1 The azido group is small, stable in biological systems, and selectively reactive with phosphines such as 1via Staudinger ligation2 and with cyclooctynes (e.g.2–5) via Huisgen [3 + 2] cycloaddition3–11 (Fig. 1). The Staudinger ligation suffers from a slow reaction rate and oxygen-sensitivity of the phosphine reagent. Thus cyclooctynes are more attractive for detection of azides on live cells. Although a number of cyclooctynes have already been reported,12 there is still a need to develop (a) more efficient syntheses, (b) faster reacting yet stable cyclooctynes and (c) cyclooctyne-based imaging agents that show minimal non-specific binding to cells and serum proteins and fast metabolic clearance in the body.13In vivo imaging studies may require large quantities of reagent. This requires efficient synthetic routes, in which there is the flexibility to generate a variety of analogues in order to tune pharmacokinetic and other properties.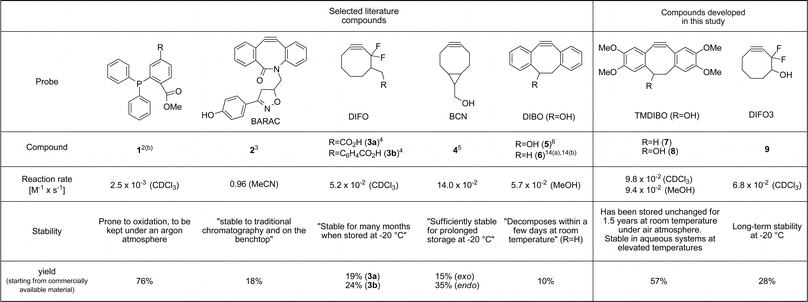 | ||
| Fig. 1 Probes for azide detection (1–5), previously synthesised cyclooctynes (6, 7) and compounds developed for this study (8, 9). | ||
In this paper we present syntheses of two new cyclooctynes, TMDIBO 8 and DIFO3 9, both of which are high yielding and easily scaled up to gram quantities. The new cyclooctynes react with azides at rates equivalent to those of other commonly used cyclooctynes and TMDIBO, in particular, shows good to excellent stability to other sets of conditions (air, acid, base, thiols). Both cyclooctynes have been used in cell-labeling studies and show much more efficient labeling than the equivalent phosphine.
Results and discussion
Boons and coworkers developed a synthesis of dibenzocyclooctyne 5, in ca. 10% yield from phenylacetaldehyde.6 Here we sought to improve ease of synthesis, total yield and stability of the final cyclooctyne. Our new tetramethoxydibenzocyclooctyne TMDIBO (8) was inspired by the work of Seitz et al.,14 who found that 7 was very stable, whereas without the methoxy groups 6 decomposed within a few days at room temperature. The synthesis of 8, shown in Scheme 1, follows the same approach as that of 5 but is much easier and higher yielding in many of the steps. Dimerisation of aldehyde 11 to yield ether 1215 is much more efficient than without the methoxy groups (95% in 2 h vs. 50% in 7 days), as is the bromination of alkene 13 (5 min vs. 12 h). It was found that the brominated product 15 is labile but can be dehydrobrominated in one pot without workup and isolation. Several bases were tried for the double dehydrobromination and 1-methylpiperazine with KOtBu gave excellent yields of 14, which was deprotected with TBAF. The overall yield of the synthesis is 57%, and flash column chromatography is only required after two of the steps.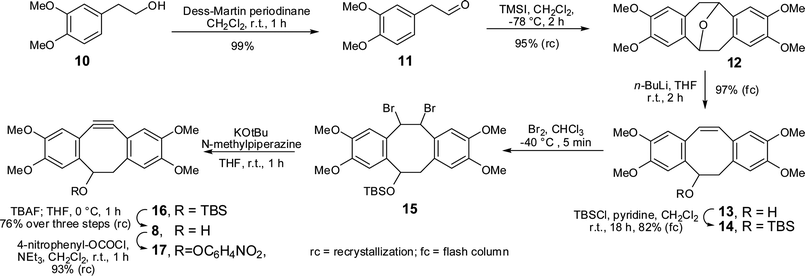 | ||
| Scheme 1 Synthesis of TMDIBO. | ||
TMDIBO is indeed remarkably stable. It has been stored in this laboratory at room temperature for 1.5 years without detectable decomposition. No oxidation of the triple bond to cyclooctane-dione could be detected, as observed in strained ring systems.16 In addition, no decomposition could be detected upon stirring in 0.3M K2CO3 in H2O–MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at reflux for 6 h. Its half life in 3M HCl/MeOH–THF (1:1:1) at 70 °C is ≈12 h. Multiple products could be detected by LCMS analysis having a mass corresponding to the addition of water. These are presumably compounds where the triple bond is hydrated to the corresponding ketones, giving two possible regioisomers, both of which could form two diastereoisomeric cyclic hemiketals. Interestingly, when TMDIBO was exposed to a 0.7 M solution of mercaptoethanol (37 °C, 12 h in either DMSO or 1:1 DMSO/PBS buffer), the strained triple bond was not affected. Instead ca. 10% of the TMDIBO was converted into a product that had a molecular weight that corresponded to replacement of the –OH group by –SCH2CH2OH, with the remainder of the TMDIBO unaffected. TMDIBO was derivatized by generating its 4-nitrophenyl carbonate 17, which in turn can be reacted with primary amines forming carbamates.
:1) at reflux for 6 h. Its half life in 3M HCl/MeOH–THF (1:1:1) at 70 °C is ≈12 h. Multiple products could be detected by LCMS analysis having a mass corresponding to the addition of water. These are presumably compounds where the triple bond is hydrated to the corresponding ketones, giving two possible regioisomers, both of which could form two diastereoisomeric cyclic hemiketals. Interestingly, when TMDIBO was exposed to a 0.7 M solution of mercaptoethanol (37 °C, 12 h in either DMSO or 1:1 DMSO/PBS buffer), the strained triple bond was not affected. Instead ca. 10% of the TMDIBO was converted into a product that had a molecular weight that corresponded to replacement of the –OH group by –SCH2CH2OH, with the remainder of the TMDIBO unaffected. TMDIBO was derivatized by generating its 4-nitrophenyl carbonate 17, which in turn can be reacted with primary amines forming carbamates.
In addition we sought to develop a synthesis of a difluorinated cyclooctyne (DIFO) that would allow the generation of large quantities of material, with minimal purification and avoiding the use of electrophilic fluorinating agents. It was thought that a DIFO might exhibit less non-specific binding to cells than TMDIBO due to its lower lipophilicity. The reported syntheses of the first- and second-generation DIFOs proceeded in ≈1% and ≈24% yield, respectively.4,7 Both rely on the rather expensive Selectfluor® to introduce the difluoromethylene group and the syntheses do not provide easy access to analogues. Therefore, the development of an efficient synthesis that would improve upon these points was investigated. A suitable precursor for our target third generation DIFO3 9 was the difluoroketone 24 synthesised by Percy and co-workers from trifluoroethanol (Scheme 2).17 The synthesis of 24 is five steps and can easily be conducted on a multigram scale, with only one step requiring column chromatography. The benzylic ether of 24 was cleanly cleaved under reductive conditions, also reducing the double bond. However, the double bond in 24 would be ideally suited for derivatisation (e.g. dihydroxylation), thereby allowing alteration of the PK properties. The resulting alcohol 25 was TBS-protected and the enol triflate 27 was formed using N-(5-chloro-2-pyridyl)triflimide. The elimination to form alkyne 28 was accomplished in good yield and the TBS-group was removed without purification of the intermediate to yield DIFO3 9. The synthesis was accomplished in 28% overall yield from trifluoroethanol, requires only four purifications by flash chromatography and use of fluorinating agents is completely avoided.
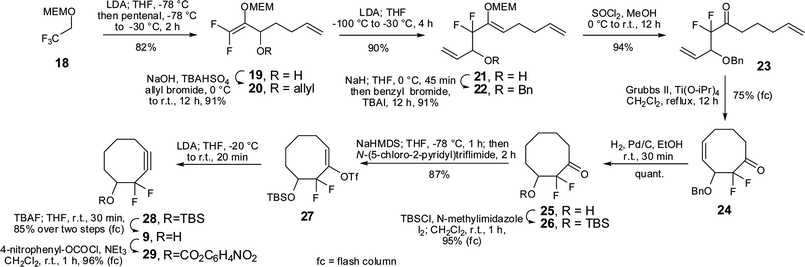 | ||
| Scheme 2 Synthesis of DIFO3. | ||
The rate constants for the [3 + 2] cycloaddition with benzyl azide were determined by 1H-NMR spectroscopy in CDCl3 at 300 K to be (9.8 ± 0.2) × 10−2M−1s−1 for TMDIBO and (6.8 ± 0.1) × 10−2M−1s−1 for DIFO3. The value for DIFO3 is comparable to that determined for DIFO2 3 (5.2 ± 0.2) × 10−2M−1s−1 (in CD3CN).4 The rate constant for TMDIBO was also conveniently measured by UV-spectroscopy in MeOH and was found to be (9.4 ± 0.4) × 10−2M−1s−1, slightly greater than the rate constant found for DIBO (5.7 ± 0.3) × 10−2M−1s−1 in the same solvent.8 Thus it is clear that the stability of TMDIBO towards decomposition has not at all compromised its rate of reaction with azides.
Next, the performance of the probes for azido glycan detection was assessed. Commonly, a two-step labeling protocol is used for glycan analysis, using biotin-derivatized probes, which are then detected using a labeled avidin.4,6 However, it would be much more practical to label the glycans in a single step. Thus, TMDIBO and DIFO3 were directly conjugated to the far-red fluorophore AlexaFluor™ 647, chosen because it can also be used for in vivo imaging. Linking Alexa Fluor 647 cadaverine to the cyclooctynes gave TMDIBO-AF64730 and DIFO3-AF64731. The corresponding PHOS-AF647 conjugate 32 was also produced (Fig. 2). A crucial quality parameter for a one-step labeling probe is its signal-to-background ratio (SBR, in this case azido-sugar treated cellsvs. untreated cells). SBR data for biotin-derivatized probes have been reported,3–8,10 whereas quantitative data for one-step labeling probes for azidoglycans are not available.
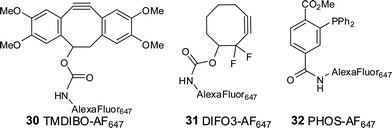 | ||
| Fig. 2 Alexa Fluor™ 647-labeled compounds. | ||
Small probes such as 30–32 should be more generally applicable for in vivo imaging than the (strept)avidin-based proteins needed to image biotinylated probes, because the small molecules should be able to diffuse to parts of the body inaccessible to proteins, which are largely restricted to the bloodstream. Furthermore the SBR achieved with biotinylated probes plus labeled (strept)avidin may be higher than for small probes because the size of the (strept)avidin may (a) prevent it reaching a probe hydrophobically bound to the plasma membrane due to obstruction by glycoproteins, etc., on the cell-surface and (b) cause a biotinylated probe-(strept)avidin complex that is weakly bound to the surface to be more easily washed away in the washing steps than a small molecule would be.
In order to compare the SBRs of our new cyclooctyne and phosphine probes, Lewis lung carcinoma (LLC) cells were cultured in the presence or absence of 50 μM peracetyl N-azidoacetylgalactosamine (Ac4GalNAz) for 24 h, resulting in azido labeling of cell surface glycans, and then washed, resuspended and exposed to the probes. The extent of the labeling was measured at various time-points by flow cytometry (Fig. 3). TMDIBO-AF647 exhibited the highest fluorescence signal at all time points with labeled cells, followed by DIFO3-AF647 and PHOS-AF647, which correlates well with their rates of reaction with benzyl azide. TMDIBO-AF647 also gave the lowest background signal with unlabeled cells, followed by PHOS-AF647 and DIFO3-AF647. This was unexpected because TMDIBO's cLogP (3.2) is higher than DIFO3's (2.7). The signal-to-background ratio for TMDIBO-AF647 (>30) is much higher than those for DIFO3-AF647 (≈10) and PHOS-AF647 (≈5). No probe-induced cellular toxicity was observed at cell labeling times typically used in the study (e.g. 15 min).
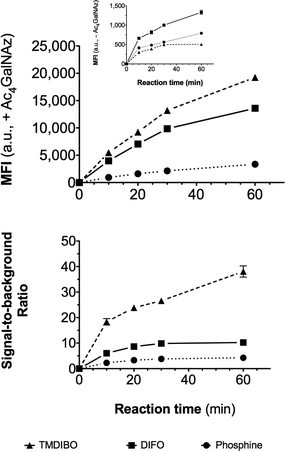 | ||
| Fig. 3 Flow cytometry data. Lewis lung carcinoma (LLC) cells were cultured with and without 50 μM Ac4GalNAz for 24 h, then incubated with a 30 μM solution of TMDIBO-AF647, DIFO3-AF647 or PHOS-AF647. Aliquots taken at various time-points were analysed by flow cytometry. Top: median fluorescence intensity (MFI) of viable cells that had been treated with Ac4GalNAz. Inset: MFI of viable cells that had not been treated with Ac4GalNAz. Bottom: Ratio of MFIs of treated vs. untreated cells. Data points were collected in triplicate and are shown as mean ± SD (error bars lie within the symbols when not visible). | ||
The labeling efficiency of TMDIBO-AF647 was confirmed by fluorescence microscopy (Fig. 4). Cells were treated as described above and automated epifluorescence laser microscopy images were analyzed for total cellular probe concentration (Fig. 4). TMDIBO-AF647 exhibited the highest signal-to-background fluorescence at 647 nm (≈15), followed by DIFO3-AF647 (≈7) and PHOS-AF647 (≈3), in agreement with flow cytometry data (Fig. 3). PHOS-AF647 showed more endocytic uptake during the incubation period of 15 min in the absence of Ac4GalNAz (see Fig. 4) and subsequent localization of the probe to perinuclear endosomes.
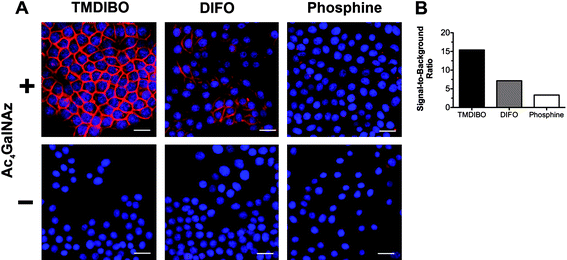 | ||
| Fig. 4 Fluorescence microscopy of LLC cell glycans (A). Cells were cultured with (+) or without (-) Ac4GalNAz (50 μM for 24 h and then incubated with a 5 μM solution of TMDIBO-AF647, DIFO3-AF647 or PHOS-AF647. Red: AF647, Blue: DAPI nuclear stain. Scale bar is 25 μm. Images were analyzed by quantitative imaging cytometry and signal-to-background ratio (B), determined from the MFI of cells cultured with (+) or without Ac4GalNAz. Signal-to-background ratios represent means of two independent measurements (ca. 10,000 events counted per well). | ||
Albumins are known to bind and remove small hydrophobic molecules from the circulation. A DIFO-based probe has recently shown significant binding to mouse serum albumin (MSA).13 The probes described here were reacted with MSA under physiological conditions and then analysed by native gel electrophoresis (Figure S3†). DIFO3-AF647 and TMDIBO-AF647 showed distinct binding to MSA (ca. 7- and 5-fold greater fluorescence than the untreated MSA) whereas PHOS-AF647 showed hardly any binding (1.1-fold increase). Prior iodoacetamide-treatment of the MSA only slightly reduced binding of DIFO3-AF647 and TMDIBO-AF647, which suggests that the majority of the bound probe is not attached to cysteine residues on MSA and is probably non-covalently bound.
Conclusions
In conclusion, efficient scalable syntheses of a new, remarkably stable dibenzocyclooctyne (TMDIBO) and a new difluorinated cyclooctyne (DIFO3) have been developed. Both cyclooctynes were tested in vitro against a phosphine reagent commonly used for azidosugar detection and it was shown that TMDIBO exhibits superior fluorescent labeling and signal-to-background ratios. Both cycloctynes showed higher levels of binding to serum albumin than the phosphine but their greater reactivity and stability would be a more important factor in many situations and give them an advantage over slower reacting, oxidation-prone phosphines. TMDIBO-based probes, in particular, show great promise for use in molecular imaging studies, including assessing cell glycosylation states in vivo and the dynamics of tumor glycosylation in relation to disease progression and response to therapy.References
- (a) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS; (b) J. M. Baskin, K. W. Dehnert, S. T. Laughlin, S. L. Amacher and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10360–10365 CrossRef CAS.
- (a) J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS; (b) E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902 CrossRef CAS.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS.
- X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS.
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS.
- E. M. Sletten, H. Nakamura, J. C. Jewett and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 11799–11805 CrossRef CAS.
- I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051–4055 RSC.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS.
- (a) G. Seitz, L. Pohl and R. Pohlke, Angew. Chem., Int. Ed. Engl., 1969, 8, 447–448 CrossRef CAS; (b) G. Seitz, L. Pohl and R. Pohlke, Angew. Chem., 1969, 81, 427–428 CrossRef.
- M. E. Jung and S. J. Miller, J. Am. Chem. Soc., 1981, 103, 1984–1992 CrossRef.
- A. Krebs and H. Kimling, Tetrahedron Lett., 1970, 10, 761–764 CrossRef.
- J. A. L. Miles, L. Mitchell, J. M. Percy, K. Singh and E. Uneyama, J. Org. Chem., 2007, 72, 1575–1587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data and NMR spectra. See DOI: 10.1039/c0sc00631a |
| This journal is © The Royal Society of Chemistry 2011 |