N-Heterocyclic carbenes versus transition metals for stabilizing phosphinyl radicals†
Olivier
Back
a,
Bruno
Donnadieu
a,
Moritz
von Hopffgarten
b,
Susanne
Klein
b,
Ralf
Tonner
b,
Gernot
Frenking
b and
Guy
Bertrand
*a
aUCR-CNRS Joint Research Chemistry Laboratory (UMI 2957), Department of Chemistry, University of California, Riverside, California 92521-0403, USA. E-mail: guy.bertrand@ucr.edu; Fax: +1 202 354 5267; Tel: +1 951 827 2719
bFachbereich Chemie, Philipps-Universitat Marburg, Hans-Meerwein-Strasse, 35032, Marburg, Germany. E-mail: frenking@staff.uni-marburg.de
First published on 18th February 2011
Abstract
It is shown that vanadium-iminato ligands are more efficient than imidazolidin-2-iminato substituents to delocalize the spin density from a phosphorus nucleus. However, the latter is stabilizing enough to allow for the isolation and characterization in the liquid and solid states of a neutral phosphinyl radical.
One significant difference between transition metals and main-group elements is that the former are generally susceptible to one-electron redox chemistry, whereas the latter are reluctant to have an odd number of electrons in their valence shell.1 An elegant demonstration of this statement can be found in the isolation by Cummins et al. of the neutral phosphinyl radical I (Fig. 1), which is stabilized by the vanadium(IV/V) redox couple.2 Indeed, despite many efforts,1,3 only a few phosphorus radicals have been isolated and characterized in the solid state4 because of their tendency to dimerize.5 It has recently been shown that, to some extent, singlet carbenes can be used as mimics for transition metal centers.6,7 In particular, stable cyclic (alkyl)(amino)carbenes (CAACs)8 and N-heterocyclic carbenes (NHCs)9 allow for the isolation of cationic paramagnetic phosphorus species II–IV.10,11 The stability of these radicals is partly due to steric factors, but more importantly to the presence of the positive charge, which prevents by electrostatic repulsion the dimerization observed for other phosphinyl radicals. Therefore, it was of interest to investigate whether singlet carbenes could also be used for the isolation of neutral phosphorus radicals, and even more importantly if they could compete with transition metal centers for stabilizing such species. Here we report the synthesis and isolation of two phosphorus radicals, which allows for a direct comparison of the electronic effects of an NHC moiety versus a metal fragment, namely V[N(Np)Ar]3 (Np=neopentyl, Ar = 3,5-Me2C6H3).
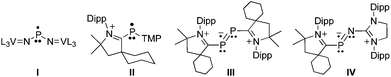 | ||
| Fig. 1 Phosphorus radicals stabilized by transition metal fragments (I) and singlet carbenes (II–IV), which have previously been structurally characterized in the solid state. L = (3,5-Me2C6H3)(Np)N; TMP = tetramethylpiperidyl; Dipp = 2,6-diisopropylphenyl. | ||
Using Cummins' radical I as a model, we targeted radical 4, in which the nitridovanadium substituents are replaced by imidazolidin-2-iminato groups.12 The cationic precursor 3 was synthesized in 3 steps starting from the free carbene 113 (Scheme 1). First the guanidine 2 was prepared in 87% yield using slightly modified known procedures.10c,14 Then in situ deprotonation of 2, followed by addition of 0.5 equivalent of PCl3 afforded salt 3 (Cl−) in a moderate yield (50%). At this stage the product contains some impurities that can be removed by anion exchange with silver triflate affording salt 3 (TfO−) in 83% yield. The 31P NMR spectrum of 3 displays a singlet at 277 ppm consistent with a dicoordinated phosphorus nucleus, and the 13C NMR spectrum shows a doublet at 158.2 ppm (JPC = 17 Hz) due to the coupling of the quaternary carbons of the five membered heterocycles with phosphorus. Lastly, the corresponding radical 4 was smoothly generated by one electron reduction of 3 using one equivalent of KC8 in THF. After work-up, 4 was obtained as a red micro-crystalline powder in 72% yield (mp. 208–211 °C).
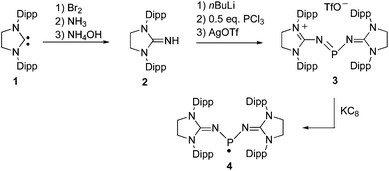 | ||
| Scheme 1 Synthesis of the phosphinyl radical 4. Dipp = 2,6-diisopropylphenyl; TfO− = trifluoromethylsulfonate. | ||
The paramagnetic nature of 4 was first indicated by silent multinuclear NMR spectra. The room temperature EPR spectrum in THF displays a large doublet at g = 2.005 due to the hyperfine coupling with the phosphorus nucleus [a(31P) = 78 G] (Fig. 2, a); no coupling with the 14N nuclei was observed. The a(31P) is in the range of values previously reported for transient phosphinyl radicals (63–100 G) where the unpaired electron mainly resides in a 3p(P) valence orbital,1,2 and is significantly larger than the value observed for the vanadium stabilized radical I (42.5 G). To gain more insights into the electronic structure of 4, a frozen solution EPR spectrum was recorded in THF at 100 K (Fig. 2, b). Simulation of the spectrum gave the following principal values for the g and 31P hyperfine coupling tensors which are aligned and display axial symmetry: gx = 2.0074, gy = 2.0062 and gz = 2.0024; Ax(31P) = Ay(31P) = 0 G and Az(31P) = 240 G. These results confirmed that the spin density is mainly localized in the phosphorus atom with 62% in the 3p(P) orbital and about 2% in the 3s(P) orbital.15 Note, that according to calculations, Cummins radical I features only 31.3% of spin density in the 3p(P) orbital,2 and about 23% over each vanadium atom.
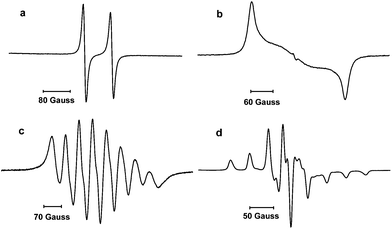 | ||
| Fig. 2 EPR spectra of 4 and 8 in a THF solution at 298 K (a and c, respectively) and in a frozen solution at 100 K (b and d, respectively). | ||
Single crystals of 4 were obtained by layering hexane on top of a THF solution allowing an X-ray diffraction study (Fig. 3, left). Radical 4 crystallizes in the C2/c space group and the asymmetric unit contains three independent molecules. In the solid state, 4 adopts a V-shape geometry with an average N1-P1-N2 angle of 97.8°. This angle is significantly more acute than the corresponding angle in the Cummins radical I (110.9°). The P1-N1 (1.658(2) Å) and P1-N2 (1.657(2) Å) bond lengths are at the lower end of the range observed for P–N single bonds. They are significantly longer than the corresponding P–N bond lengths in I (1.63 Å and 1.61 Å), in line with the weaker spin delocalization in 4.
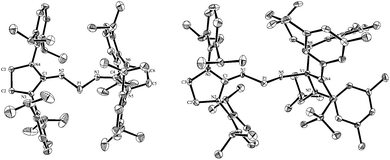 | ||
| Fig. 3 Molecular views (with 50% thermal ellipsoids) of one of the three molecules of 4 (left) and 8 (right) in the solid state. For clarity, the hydrogen atoms have been omitted. Selected bond lengths (Å) and angles (deg). 4: P1-N2, 1.657(2); P1-N1, 1.658(2); N1-C4, 1.272(4); N2-C1, 1.277(4); N2-P1-N1, 96.75(13). 8: P1-N5, 1.572(5); P1-N1, 1.634(5); N1-C1, 1.286(7); V1-N5, 1.806(4); V1-N6, 1.902(5); V1-N7, 1.898(4); N1-P1-N5, 109.5 (3). | ||
In order to directly compare the stabilizing ability of the imidazolidin-2-iminato substituent with the nitridovanadium group, we undertook the synthesis of the mixed substituted phosphinyl radical 8 (Scheme 2). The dichlorophosphine 5, readily prepared from 2, was reacted in THF with the already reported vanadium nitride anion 6,16 affording the chlorophosphine 7 in 73% yield. The 31P NMR spectrum of 7 displays a broad singlet at 185 ppm. This relatively high field chemical shift shows that unlike 3 the chlorine stays coordinated to the phosphorus atom of 7. This suggests that the imidazolidin-2-iminato fragment is a better electron releasing group than the nitridovanadium metalloligand. Because of the chirality at the phosphorus center, each pair of methylene protons in the neopentyl moieties are diastereotopic resulting in two doublets at 4.54 and 4.44 ppm in the 1H NMR spectrum. Similarly, the isopropyl groups of the Dipp groups give rise to two sets of signals in the 1H and 13C NMR spectra.
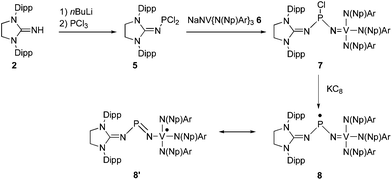 | ||
| Scheme 2 Synthesis of the mixed NHC-Vanadium phosphinyl radical 8, and its most representative resonance form 8′ (Dipp = 2,6-diisopropylphenyl, Np = Neopentyl, Ar = 3,5-Me2C6H3). | ||
Reduction of 7 with one equivalent of KC8 in THF afforded after work-up the radical 8 as a dark red powder in 85% yield (mp. 98–102 °C). The room temperature EPR spectrum in THF displays an eight lines pattern (g = 1.981) due to the hyperfine coupling with the 51V nucleus [a(51V) = 58 G] (Fig. 2, c). According to the simulation, the coupling with the 31P nucleus is weak and results only in a slight broadening of the lines. The frozen solution EPR spectrum was recorded at 100 K in THF (Fig. 2, d). Simulation of this spectrum allowed the determination of the principal values of the g and hyperfine tensors which are aligned: gx = 1.9726, gy = 2.0048 and gz = 1.9583; Ax(51V) = Ay(51V) = 30 G and Az(51V) = 121 G; Ax(31P) = Ay(31P) = 7 G and Az(31P) = 12 G. Note that these values are quite close to those reported for the vanadium(IV) complex V(NEt2)4.17 Therefore, according to these values,15 and considering the fact that the 51V hyperfine coupling tensor displays axial symmetry, the spin density is mainly localized at the vanadium (67%) and very lightly in the phosphorous 3p orbital (1%) as well as on the NHC fragment.
The structure of radical 8 was unambiguously confirmed by an X-ray diffraction analysis performed on a single crystal grown by slow evaporation of an ether solution of 8 at room temperature (Fig. 3, right). In the solid state 8 adopts a V-shape geometry with a N5-P1-N1 angle of 109.5(3)°, which is broader than in 4 (96.75(13)°), and comparable to that observed for I (110.9°). The value of the P1-N5 bond length (1.572(5) Å) lays in the range for PN double bonds, and the V1-N5 bond length (1.806(4) Å) is even longer than the corresponding value in I (1.72 Å).
DFT calculations of the real compounds I, 4 and 8 have been carried out to provide more insight into their electronic structure.18 The calculated spin densities for I are in rough agreement with the previous calculations by Cummins which were carried out for the model compound Im, which was restricted to C2 symmetry and where the large aryl and bulky alkyl groups were replaced by phenyl and methyl, respectively.2 The orbital analysis suggests that there is a rather large contribution of the phosphorous orbitals to the excess spin density (+0.42 e) and equally large contributions of the 3d orbitals of each vanadium atom (+0.43 e/+0.44 e) to the SOMO (Fig. 4a). In the mixed compound 8, the single vanadium atom clearly possesses the largest atomic spin density of all atoms which is even slightly larger than the spin density at both vanadium atoms in I while the excess spin density at P is only half as large as in I (Fig. 4c). In compound 4, the phosphorous atom carries a much larger part of the spin density (+0.68 e) while the carbon atoms of the adjacent NHC rings only exhibit approximately 0.1 e excess spin density (Fig. 4b). The vanadium atom thus appears to be much better suited for carrying excess spin density than the NHC rings. Note that the nitrogen atoms of 4 carry a slight excess of beta electron density (negative atomic spin density values) thus confirming that they do not play a role in stabilizing the radicals.
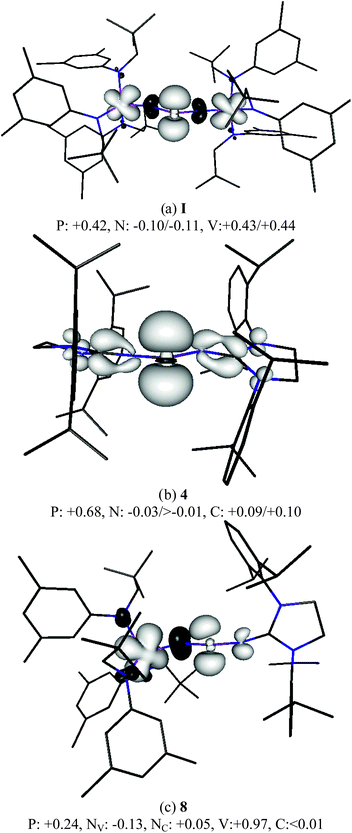 | ||
| Fig. 4 Spin density (BP86/TZVPP//BP86/SVP, isosurfaces at 0.004 and −0.004 a.u) of I, 4 and 8. Mulliken atomic spin densities per atom given in electrons for the atoms of the central moieties. | ||
In order to estimate the effect of the substituents (NHC, L3V) on the radical stability of I, 4 and 8, the energies of the isodesmic reactions RR′P-H + PH2 → RR′P + PH3 (RR′P = I, 4 and 8) were calculated. At the BP86/TZVPP//BP86/SVP level of theory, the value for the vanadium complex I is −26.3 kcal mol−1, which is in good agreement with the data reported for the model vanadium compound (−24 kcal mol−1).2 For the mixed compound 8, a comparable value was found (−26.5 kcal mol−1), but the exothermicity significantly decreases for the bisNHC derivative 4 (–18.1 kcal mol−1).
According to these experimental and computational results, the paramagnetic species 4 is better described as a phoshorus-center radical, with little delocalization over the imidazolidin-2-iminato substituents. In contrast, 8 is best represented by the resonance structure 8′, which corresponds to a vanadium(IV) complex containing an imidazolidin-2-iminatophosphinimide ligand (Scheme 2). Lastly, the spin density distribution in compound I lays in between 4 and 8 since both the phosphorous and vanadium centers possess large spin densities.
Conclusions
These results as a whole demonstrate that the vanadium-iminato ligand is more efficient than the imidazolidin-2-iminato substituent to delocalize the spin density from the phosphorus nucleus. However, the latter is efficient enough to allow the isolation and characterization in the liquid and solid states of a phosphinyl radical. It is quite likely that the use of imidazolidin-2-iminato groups and related species will permit the isolation of a variety of main group element centered radicals.Acknowledgements
Financial support from the NSF (CHE-0924410), RHODIA Inc, and DFG is gratefully acknowledged. Thanks are due to Dan Borchardt for the EPR part of this paper.Notes and references
- P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS.
- P. Agarwal, N. A. Piro, K. Meyer, P. Muller and C. C. Cummins, Angew. Chem., Int. Ed., 2007, 46, 3111 CrossRef CAS.
- For reviews on phosphorus radicals, see: (a) A. Armstrong, T. Chivers and R. T. Boere, ACS Symp. Ser., 2006, 917, 66 CAS; (b) S. Marque and P. Tordo, Top. Curr. Chem., 2005, 250, 43 CAS; (c) M. Geoffroy, Recent Res. Dev. Phys. Chem., 1998, 2, 311 Search PubMed.
- (a) M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., Int. Ed., 2009, 48, 2600 CrossRef CAS; (b) S. Ito, M. Kikuchi, M. Yoshifuji, A. J. Arduengo, T. A. Konovalova and L. D. Kispert, Angew. Chem., Int. Ed., 2006, 45, 4341 CrossRef CAS; (c) A. Armstrong, T. Chivers, M. Parvez and R. T. Boere, Angew. Chem., Int. Ed., 2004, 43, 502 CrossRef CAS.
- (a) M. J. S. Gynane, A. Hudson, M. F. Lappert, P. P. Power and H. Goldwhite, J. Chem. Soc., Chem. Commun., 1976, 623 RSC; (b) S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, Chem. Commun., 2000, 2045 RSC; (c) S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. MacDonald, R. J. Wiacek, A. Voigt, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, J. Am. Chem. Soc., 2001, 123, 9045 CrossRef CAS; (d) J. P. Bezombes, K. B. Borisenko, P. B. Hitchcock, M. F. Lappert, J. E. Nycz, D. W. H. Rankin and H. E. Robertson, Dalton Trans., 2004, 1980 RSC.
- See for examples: (a) G. D. Frey, V. Lavallo, B. Donnadieu and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS; (b) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS; (c) Y. Wang and G. H. Robinson, Chem. Commun., 2009, 5201 RSC; (d) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 CrossRef CAS; (e) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295 CrossRef CAS; (f) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. von, R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069 CrossRef CAS; (g) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaeffer III, P. von, R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970 CrossRef CAS.
- For reviews, see: (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS; (b) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389 RSC; (c) C. A. Dyker and G. Bertrand, Nat. Chem., 2009, 1, 265 CrossRef.
- For a review, see: M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 Search PubMed.
- For a recent review, see: F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 Search PubMed.
- (a) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262 CrossRef CAS; (b) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS; (c) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930 CAS.
- It has also been shown that singlet carbenes considerably enhance the stability of boryl radicals. (a) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256 CrossRef CAS; (b) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256 CrossRef CAS; (c) J. C. Walton, M. Makhlouf Brahmi, L. Fensterbank, E. Lacôte, M. Malacria, Q. Chu, S.-H. Ueng, A. Solovyev and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 2350 CrossRef CAS; (d) T. Matsumoto and F. P. Gabbai, Organometallics, 2009, 28, 4252 CrossRef CAS.
- For recent papers dealing with imidazolin-2-iminato as ligands for transition metals, see: (a) A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156 CrossRef CAS; (b) A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435 CrossRef CAS; (c) B. Haberlag, X. A. Wu, K. Brandhorst, J. Grunenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2010, 16, 8868 CrossRef CAS; (d) T. Gloge, D. Petrovic, C. Hrib, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2009, 4538 CrossRef; (e) T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462 CrossRef CAS; (f) S. Beer, K. Brandhorst, C. G. Hrib, X. A. Wu, B. Haberlag, J. Grunenberg, P. G. Jones and M. Tamm, Organometallics, 2009, 28, 1534–1545 CrossRef CAS.
- A. J. Arduengo III, R. Krafczyk and R. Schmutzler, Tetrahedron, 1999, 55, 14523 CrossRef CAS.
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793 CrossRef CAS.
- J. R. Morton and K. F. Preston, J. Magn. Reson., 1978, 30, 577 CAS.
- J. K. Brask, V. Durà-Vilà, P. L. Diaconescu and C. C. Cummins, Chem. Commun., 2002, 902 RSC.
- C. E. Holloway, F. E. Mabbs and W. R. Smail, J. Chem. Soc. A, 1968, 2980 RSC.
- Geometry optimisations without symmetry constraints were carried out at the BP8619/def2-SVP20 level of theory for the real systems which are presented here and for model compounds which have smaller substituents. The latter systems have been verified as minima by calculating the Hessian matrices. Frequency calculations of the real systems could not be carried out due to the size of the molecules but they can be assumed to be energy minima as well. Spin densities and population analyses were derived via single-point calculations with the def2-TZVPP20 basis set. The program packages TurboMole21 and Gaussian0322 were employed. The full ref. 22 and the cartesian coordinates of the calculated compounds are given in the supporting information.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- F. Weigend and R. Ahlrichs, PhysChemChemPhys, 2005, 7, 3297 RSC.
- TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com Search PubMed.
- M. J. Frisch et al. , Gaussian, Inc., Gaussian 03, Revision E.01, Wallingford CT, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, physical and spectroscopic data for all new compounds, and computational details. CCDC 808620–808621. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00027f |
| This journal is © The Royal Society of Chemistry 2011 |