Catalytic functions of cubane-type M4S4 clusters†
Hidetake
Seino
*a and
Masanobu
Hidai
*b
aInstitute of Industrial Science, The University of Tokyo, Komaba, Meguro-ku, Tokyo, 153-8505, Japan. E-mail: seino@iis.u-tokyo.ac.jp; Fax: +81 3 5452 6361; Tel: +81 3 5452 6362
bDepartment of Materials Science and Technology, Faculty of Industrial Science and Technology, Tokyo University of Science, Noda, Chiba 278-8510, Japan. E-mail: hidai@rs.noda.tus.ac.jp
First published on 15th February 2011
Abstract
Catalysis by the cubane-type Fe4S4 cluster has been found in metalloproteins such as biotin synthase, aconitase, and IspH, whereas some synthetic M4S4 clusters show catalytic activities for biologically related reactions or various types of organic transformations. This minireview summarizes recent progress in the chemistry of catalytic functions of cubane-type M4S4 clusters.
 Hidetake Seino | Hidetake Seino received his doctorate in 1997 from the University of Tokyo under the supervision of Professor M. Hidai, where he studied the synthesis of organonitrogen compounds by using transition metal–dinitrogen complexes. Then he joined the group of Professor Y. Mizobe at the Institute of Industrial Science, the University of Tokyo, investigating the chemistry of metal–sulfide clusters and the conversion of small molecules by using transition metal complexes. His current research interests focus on development of new catalytic systems by learning from the mechanisms of metalloenzymes. |
 Masanobu Hidai | Masanobu Hidai, born in 1940, received his doctorate from the Department of Industrial Chemistry at the University of Tokyo in 1968. He then joined the staff and became Full Professor in 1986. From 1976 to 1977, he spent time at the University of Sussex, UK, working with Professor J. Chatt. He received the award of the Chemical Society of Japan in 1996. After his retirement from the University of Tokyo in 2000, he continued his research at Tokyo University of Science until 2006. His research interests include the chemistry of nitrogen fixation and organic synthesis catalyzed by transition metal complexes. |
Introduction
The cubane-type M4S4 structure, in which four metal atoms (Mn+) and four μ3-sulfido ligands (S2−) are alternately occupying vertices of a cubic-like framework, is one of representative morphologies of metal sulfido clusters and has appreciable thermodynamic stability. These types of clusters are active for multi-step redox and coordination change at the metal sites, while the fairly robust cubane-type cores are retained in these processes. The cubane-type Fe4S4 cluster is often observed in metalloproteins such as ferredoxin, hydrogenase, and the Fe protein of nitrogenase, where the major function of the cluster is to facilitate electron transfer.1 However, the Fe4S4 unit in aconitase is well known to catalyze the stereospecific interconversion of citrate and isocitrate, which represents a vital metabolic pathway in the tricarboxylic acid (TCA) cycle. Further, an Fe4S4 cluster is combined with another metal unit by a bridging cysteinyl ligand to compose the active sites in metalloenzymes such as sulfite reductase,2 [FeFe] hydrogenase (H-cluster),3 and acetyl coenzyme A synthase (A-cluster).4 Recent advances in inorganic synthesis have now enabled the synthesis of a variety of homonuclear and heteronuclear cubane-type M4S4 clusters containing not only the first-row transition metals, but also the second- and third-row transition metals.5–9 Their structures and reactivities have extensively been studied partly in relevance to biological functions of metalloproteins.5–7 Furthermore, unique catalysis by cubane-type clusters containing noble metals in particular, which are not found in metalloproteins, has also been explored.9,10 This minireview is the first attempt to focus on catalytic functions of cubane-type M4S4 clusters covering a wide field of research, including bioinorganic, inorganic, organometallic, organic, and electro- chemistry.1. Biological organic transformations catalyzed by the cubane-type Fe4S4 cluster in enzymes
The biological Fe4S4 clusters that play a role in electron transfer are generally coordinated by four cysteine residues in proteins. On the other hand, most of the Fe4S4 clusters catalyzing chemical transformations possess a common structure, in which three iron atoms are bound to cysteinyl ligands, and the remaining fourth iron atom serves as the coordination site of substrates (Chart 1).11 Several types of catalytic functions have been revealed for such Fe4S4 active sites.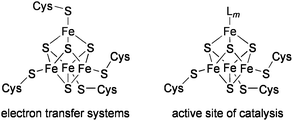 | ||
| Chart 1 Biological Fe4 S4clusters. | ||
The radical SAM superfamily is a group of enzymes that utilize S-adenosylmethionine (SAM) to initiate diverse radical mediated reactions (Chart 2).12 About 3000 enzymes of this family have been found, and some of them have been determined by X-ray crystallographic structure analysis, e.g.lysine 2,3-aminomutase (LAM), an enzyme for biosynthesis of molybdopterin (MoaA), biotin synthase (BioB), and coproporphyrinogen oxidase (HemN). Extensive spectroscopic studies suggest that the enzymatic mechanisms of this superfamily involve essentially common initial steps (Scheme 1). A [Fe4S4]+cluster coordinates SAM at the unique iron subsite and transfers one electron to SAM. This causes cleavage of the C–S bond to generate methionine (Met) and 5′-deoxyadenosyl radical (DOA·), and the latter mediates various types of radical reactions. In the reaction of LAM, this radical abstracts a β-hydrogen of the α-lysine that is in a form of Schiff base with pyridoxal 5′-phosphate (PLP), and the generated carbon radical rearranges with migration of the nitrogen to the β-carbon. The resulting α-C radical is donated a hydrogen from DOAH to give the β-lysine–PLP. Reaction of BioB consumes two equivalents of SAM to cleave two unreactive C–H bonds, and the second iron-sulfur center [2Fe-2S] supplies a sulfur atom to the activated substrate. It is proposed that the sulfur atom of the formed Met coordinates to the unique iron when SAM acts as a catalyst (e.g.LAM and MoaA). In contrast, association of the Met sulfur with a cluster sulfide is suggested for the enzymes that use SAM as a substrate (e.g. BioB and HemN).
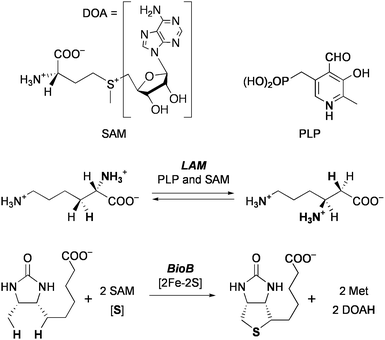 | ||
| Chart 2 Examples of reactions catalyzed by radical SAM enzymes. | ||
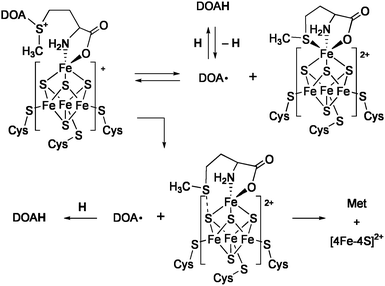 | ||
| Scheme 1 Two mechanisms for the reductive cleavage of SAM. | ||
Aconitase catalyzes the stereospecific interconversion of citrate and isocitratevia cis-aconitate, comprising a part of the TCA cycle.13 X-ray crystal structure determinations of aconitase in the substrate-free form and with bound isocitrate have shed light on critical aspects of the catalytic mechanism.14Scheme 2 shows the proposed mechanism for the conversion of isocitrate to citratevia cis-aconitate at the active site of aconitase. In the resting state, the unique iron atom is bound by a hydroxide ion. In the presence of the substrate, the hydroxide ion is protonated, and the resulting water ligand and the substrate are both coordinated to the unique iron. Consequently, the geometry of the iron changes from four-coordinate to six-coordinate. The substrate is then dehydrated to form cis-aconitate at this site through attack by Ser-642 alkoxide and His-101 imidazolium. Citrate is formed via the reorientation of cis-aconitate to the other binding mode (180° rotation) followed by rehydration. The reaction proceeds via a nonredox process, and the unique iron center just acts as a Lewis acid. There are considerable numbers of enzymes that contain the active sites closely related to aconitase in the hydro-lyase family.
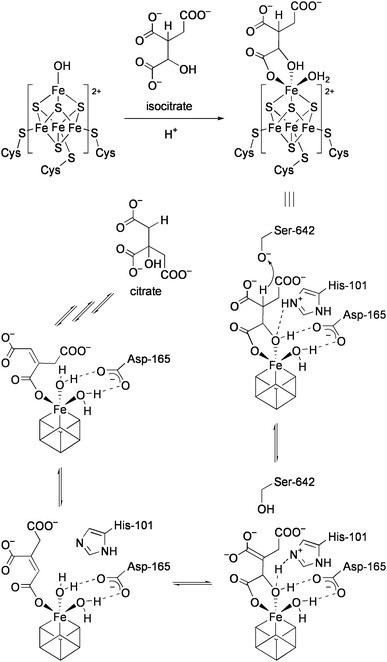 | ||
| Scheme 2 Mechanisms for the conversion of isocitrate to citratevia cis-aconitate. | ||
An Fe4S4 cluster containing protein, called IspH, which is found in pathogenic microorganisms, catalyzes the formation of isoprenoid building blocks, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) by the 2H+/2e−reduction of (E)-1-hydroxy-2-methyl-2-butenyl 4-diphosphate (HMBPP). Quite recently, a novel bioorganometallic mechanism for IspH catalysis has been proposed on the basis of X-ray structure analysis15 as well as detailed spectroscopic investigations of action and inhibition of IspH.16 The proposed mechanism of action of IspH is shown in Scheme 3. HMBPP initially binds to the unique iron in a [Fe4S4]2+ cluster to form a hydroxyl complex. Reduction of the cluster by one electron gives a π-complex of HMBPP, which is followed by protonation to remove the 4-OH as H2O and afford an η1-allyl complex. The cluster is then reduced by the second electron to form an η3-allyl complex. Finally, protonation at C2 or C4 gives rise to the formation of IPP or DMAPP, respectively. It has been demonstrated that IspH is effectively inhibited by alkynyl diphosphates, which probably form more stable π-complexes than alkenyl compounds. Another organometallic mechanism has quite recently been proposed for action and inhibition of the Fe4S4 isoprenoid biosynthesis protein, called GcpE, which catalyzes the 2H+/2e−reduction of 2-C-methyl-D-erythritol-2,4-cyclo-diphosphate (MEcPP) to HMBPP.17 It is supposed that MEcPP is converted at the unique iron atom into the intermediate having a ferraoxetane structure, which is then deoxygenated to an η2-alkene complex of HMBPP. An alkyne analogue of HMBPP has been shown to inhibit GcpE. With regard to binding and conversion of MEcPP, however, the possibility of alternative pathways has been suggested, and the detailed mechanism is inconclusive at present.18
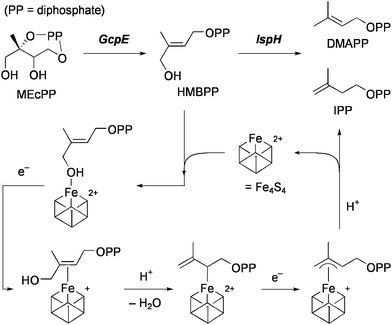 | ||
| Scheme 3 Two steps in the isoprenoid biosynthesis and the proposed IspH mechanism. | ||
2. Catalysis by synthetic Fe4S4 clusters as biological models and its applications
In relevance to the redox functions and the above catalytic roles of the metalloproteins containing the cubane-type Fe4S4 clusters, many studies have been performed by using the synthetic analogues [Fe4S4(SR)4]n− (n = 1–3) since the first preparation by Holm and co-workers (Chart 3, top).19 They demonstrated the reductive cleavage of sulfonium cations by [Fe4S4(SR)4]3− as the reactivity analogue of SAM enzymes.20,21 The Fe4S4 clusters, whose iron subsites are differentiated in the ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, were also developed by use of a tridentate thiolate ligand bound to the three iron atoms. The unique iron atom in these site-differentiated clusters binds various ligands (hydroxide, carboxylates, carbon donors, etc.) and adopts four-, five-, and six-coordinate geometries via the site-specific substitution reactions (Chart 3, bottom).21,22 Thus, these synthetic clusters provide good structural models for the active site clusters in enzymes including aconitase. Catalytic hydrolysis of aryl esters by [Fe4S4(SPh)4]2− (1a) was examined by another group in relation to the function of aconitase.23
:1, were also developed by use of a tridentate thiolate ligand bound to the three iron atoms. The unique iron atom in these site-differentiated clusters binds various ligands (hydroxide, carboxylates, carbon donors, etc.) and adopts four-, five-, and six-coordinate geometries via the site-specific substitution reactions (Chart 3, bottom).21,22 Thus, these synthetic clusters provide good structural models for the active site clusters in enzymes including aconitase. Catalytic hydrolysis of aryl esters by [Fe4S4(SPh)4]2− (1a) was examined by another group in relation to the function of aconitase.23
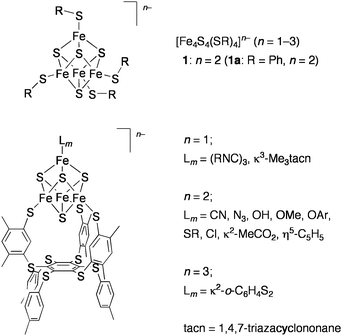 | ||
| Chart 3 Synthetic Fe4 S4clusters. | ||
Similarly to the biological role of [Fe4S4(S-Cys)4]n−, the synthetic Fe4S4 clusters are utilized as catalyst precursors in electron transfer reactions. The groups of Tezuka and Hidai reported that controlled-potential electrolysis in the presence of [Fe4S4(SCH2Ph)4]2− resulted in reduction of carbon dioxide at more positive potentials (∼ −1.7 V vs.SCE) than those without catalysts (–2.4 V vs.SCE).24Formate was predominantly produced instead of oxalate. Analogous reactions catalyzed by the Fe4S4 clusters bearing a macrocyclic tetrathiolate ligand have been reported.25 Other electrochemical reductions that convert nitrogenase substrates are described in the next section.
Redox reactions between electron-donor and -acceptor molecules are facilitated by 1 and analogues as summarized in Chart 4. In the presence of a catalytic amount of 1a, thiols are smoothly oxidized to disulfide by O2, quinones, and other organic electron acceptors.26Reduction of organic nitro compounds with thiols to give amines was demonstrated by this method.27,28Oxidation of benzoin with benzoquinone was examined by using analogues of 1 with bulky thiolate ligands or cysteine-containing peptides.29 It has been proposed that the oxidized Fe4S43+ core formed viaelectron transfer to quinone binds benzoin at the iron subsite by replacing the thiolate ligand and is reduced to the Fe4S4+ state concurrent with the fomation of benzil. The agglomerates, in which the Fe4S4 cores were cross-linked by dithiolate ligands had similar catalytic activity.30
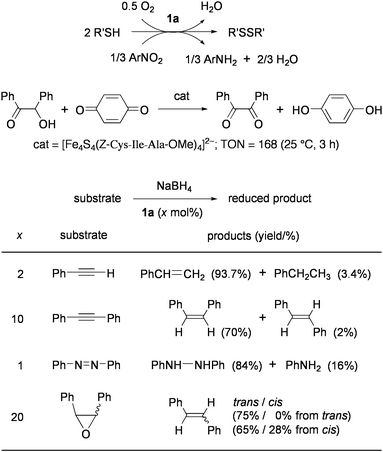 | ||
| Chart 4 Redox-reactions catalyzed by synthetic Fe4S4 clusters. | ||
When 1a and NaBH4 are used as the catalyst and the electron donor respectively in protic media, phenylacetylene and diphenylacetylene are hydrogenated predominantly to styrene and cis-stilbene respectively, and azobenzene is reduced to hydrazobenzene and aniline.31,32 This method has also been applied to deoxygenation of oxiranes into olefins, which is assumed to be the crucial function of GcpE enzyme (vide supra).33 Stoichiometric studies on reduction of proton and acetylene to H2 and ethylene, respectively, have confirmed that the reduced cluster of Fe4S4+ core acts as the one electron donor and that a thiolate ligand in this cluster presumably dissociates to open the iron subsite for substrate binding.34–36
Treatment of [Fe4S4Cl4]2− with phenyllithium under H2 gave a catalyst for hydrogenation of alkenes, which operated under 1 atm of H2 at room temperature.37 Addition of a mixture of PhSH and PhSSPh after reaction allowed recovery of cluster species as 1a. In contrast, treatment of 1 with phenyllithium exerted no such hydrogenation activity under H2.
3. Nitrogenase-related transformations catalyzed by cubane-type MFe3S4 clusters (M = Mo, V, Fe)
Nitrogenase is an enzyme, which converts atmospheric N2 into ammonia by use of electrons and protons. Three types of nitrogenases are known, which possess MoFe, VFe, or Fe-only sulfide clusters at their active sites, and the efficiencies of the enzymes decrease in this order. The recent single crystal X-ray analysis of the Mo-containing nitrogenase has revealed that the active site (FeMo-co) is an unprecedented MoFe7S9X cluster with two incomplete cubane-type MoFe3S3 and Fe4S3 units bridged by three μ2-sulfido ligands and the interstitial μ6-X atom (X = N, O, or C) (Chart 5).38 In the late 1970s, however, the active site of the nitrogenaseMoFe protein was presumed to be a cubane-type MoFe3S4 cluster on the basis of Mo EXAFS analysis of the MoFe protein.39 A similar cubane-type VFe3S4 core has been also suggested for the active site of the V-containing nitrogenase,40,41 although its crystallographic structure is not yet available. In this context, intensive studies had been focused on the synthesis and reactivity of cubane-type MFe3S4 (M = Mo, V) clusters (Chart 6).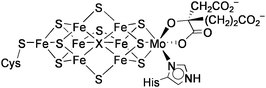 | ||
| Chart 5 Structure of FeMo-co. | ||
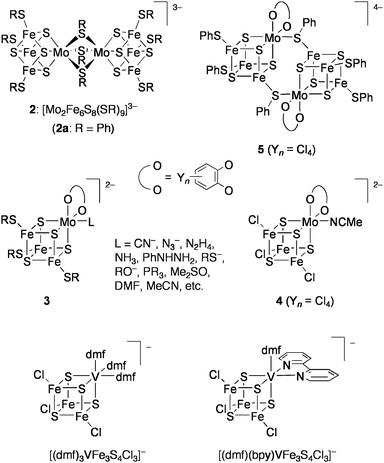 | ||
| Chart 6 Synthetic cubane-tube MoFe3S4 and VFe3S4 clusters investigated for nitrogenase-related catalysis. | ||
The groups of Holm and Garner almost simultaneously reported synthesis of the first synthetic cubane-type MoFe3S4 clusters in the dimeric forms such as [Mo2Fe6S8(SR)9]3− (2), in which the Mo sites of two MoFe3S4 cores were connected by bridging thiolate ligands.42,43 The monomeric clusters [(cat)(L)MoFe3S4(SR)3]2− (3: cat = catecholate; L = 2e donors) were prepared later44 and proven to coordinate various pseudosubstrates of nitrogenase, such as CN−, N3−, N2H4, NH3, and MeCN, as L at the Mo site.45 These clusters undergo reversible 1e and 2e (only for 2) reductions, and the reduced clusters revert to the initial oxidation level on treatment with proton sources (e.g., PhSH or Et3NH+) accompanied with formation of H2.46Proton reduction is known as one of the functions of nitrogenase.
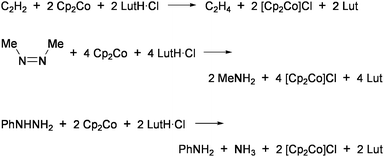
Coucouvanis and co-workers demonstrated nitrogenase-related transformations by using the analogue of 3, [(Cl4-cat)-(MeCN)MoFe3S4Cl3]2− (4: Cl4-cat = tetrachlorocatecholate).47 The reduction of hydrazine to ammonia in the system utilizing cobaltocene (Cp2Co) and 2,6-lutidinium chloride (LutH·Cl) as external sources of electrons and protons, respectively (Eq 1), was catalyzed by 4 at ambient temperature in MeCN.48–50 This cluster also promoted the disproportionation reaction of hydrazine (Eq 2) at a rate slower than the above reduction by one order of magnitude. The iron-only cluster [Fe4S4Cl4]2− was inactive for these reactions. Reduction of hydrazine is considered to proceed at the single Mo site and is inhibited remarkably by PEt3 and CO, which bind tightly to the Mo site. Although several double-cubane clusters containing a hydrazine ligand as the bridging unit between the Mo atoms were isolated,51 these showed quite poor activities. An analogous cluster [(Hcitr)MoFe3S4Cl3]3− (Hcitr = κ3-citrate trianion), which was prepared in relevance to the homocitrate-binding Mo site in FeMo-co, exhibited an even higher catalytic rate for hydrazine reduction (about 80 cycles in the initial 5 min under certain conditions) than 4 but had no activity for disproportionation. It is presumed that the coordination site for hydrazine is formed via protonation at the tridentate Hcitr ligand. Similar catalytic activities were also observed for other MoFe3S4 clusters binding carboxylate ligands at the Mo site.52
| N2H4 + 2Cp2Co + 4LutH·Cl→2NH+4 + 2[Cp2Co]Cl + 4Lut | (1) |
| 3N2H4→4NH3 + N2 | (2) |
Employment of Cp2Co and LutH·Cl as sources of electrons and protons, respectively, in the presence of 4 as a catalyst has further enabled the reduction of acetylene into ethylene,53cis-dimethyldiazene into methylamine,54 or phenylhydrazine to aniline and ammonia (Chart 7).49Methylhydrazine was converted to methylamine and ammonia, though in competition with the reduction of proton to evolve H2.48 The vanadium containing cubane-type clusters [(L)3VFe3S4Cl3]− ((L)3 = (dmf)3, (dmf)(bpy): dmf = N,N-dimethylformamide; bpy = 2,2′-bipyridyl: Chart 6)55 were also shown to be catalytically effective for the reduction of hydrazine or phenylhydrazine.48,56
 | ||
| Chart 7 Reductions relevant to nitrogen fixation catalyzed by 4. | ||
Cluster 3 (cat = Cl4-cat; L = Me2CO; R = p-C6H4-n-C8H17) catalyzed the reduction of n-C5H11N3 by Na2S2O4 in aqueous Triton X-100 micellar solutions, giving equal amount of n-C5H11NH2 and N2 (Eq 3, R = n-C5H11).57 Addition of methyl viologen to these system not only increased reaction rates but also gave ammonia and hydrazine, whose production required larger numbers of electrons per substrate. The iron cluster 1 (R = p-C6H4-n-C8H17) catalyzed only Eq 3 under the same conditions.
| RN3 + 2e− + 2H+→RNH2 + N2 | (3) |
Tanaka and co-workers extensively investigated the catalytic activities of the cubane-type MoFe3S4 and Fe4S4 clusters for nitrogenase-related transformations under controlled potential electrolysis conditions (Hg working electrode). The double cubane-cluster 2a was effective for the reduction of hydrazine into ammonia in methanol/THF solution at the potential that attained 2e reduction of 2a (–1.25 V vs.SCE).58Reduction of proton to form H2 competitively took place (vide supra) only in a small ratio (∼3%). The iron-only cubane cluster 1a had higher catalytic activity for H2 evolution than 2a59 and exhibited inferior selectivity for reduction of hydrazine against that of proton (44:56). The proportion of H2 evolution became greater in aqueous media for both 1a and 2a, especially at low pH values. Acetylene was also preferentially reduced in these systems into ethylene.59,60 In contrast to single cubane clusters 3 and 4, double cubane clusters 2 are proposed to bind substrates not at the Mo atom but at the Fe subsite that has an easily dissociable terminal thiolate ligand. Observations that acetylene36 or methyl azide61 coordinate to the iron subsite in the 1e-reduced species of 2a were claimed. Reduction of CH3NC by electrolysis in the presence of 1a and 2a gave CH3NH2, CH4, C2H6, and a small amount of C2H4.62 These hydrocarbons are considered to originate from the isocyanide carbon, and it is explained that C2H6 is produced via the insertion of CH3NC into the CH3–metal bond that is formed at an intermediary stage of reduction. Acetonitrile was also reduced to C2H6, C2H4, and NH3 in these systems. Electrolysis with 2a converted CH3N3 into CH3NH2 and N2via a 2e-transfer process (Eq 3, R = CH3) with nearly 100% current efficiency at high substrate concentration, while hydrazine and ammonia were also formed in dilute solution as multielectron reduction products.61,63
In similar systems using 1 or 2, N3− was transformed into N2 and ammonia in about a 2:1 ratio, concomitantly forming large amounts of H2 (more than 10-fold of NH3) and a little hydrazine.64 Depression of H2 evolution and improvement of the ammonia yield was achieved by using the working electrodes, which were prepared by modifying glassy-carbon plates with 2a.65 Superiority of this electrode in multielectron reduction of HOCH2CH2N3 was demonstrated in aqueous solution to give large amounts of ammonia with high catalytic rates (Eq 4).61,63Electrodes modified with 1a and the double-cubane cluster [{(Cl4-cat)MoFe3S4(SPh)2}2(μ2-SPh)2]4− (5: Chart 6), which was regarded as the dimer form of 3, were also tested for the reduction of HOCH2CH2N3 in water (Eq 5).66
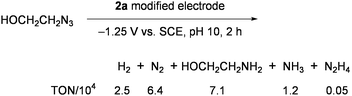 | (4) |
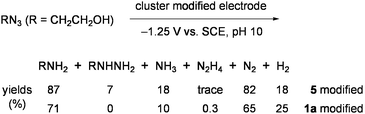 | (5) |
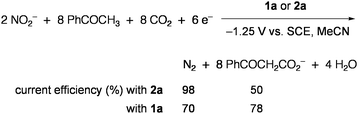
Electrolysis using the electrode modified with 2a or 5 selectively reduced NO2− and NO3− to ammonia at −1.25 V vs.SCE in aqueous solution.67 When a CO2-saturated MeCN solution containing NO2−, PhCOCH3, and 3Å molecular sieves was electrolyzed in the presence of 2a, PhCOCH3 acted as the proton source in reduction of NO2− into N2 and was subsequently carboxylated to give PhCOCH2CO2− (Eq 6).68 Similar activity was also observed for 1a, and other organic compounds having active hydrogens (pKa = 18–21) could replace PhCOCH3. The CO2 fixation via 2e reduction was performed in the system that utilized thioesters R′COSEt (R′ = CH3, C2H5, Ph) as the CO2 acceptor and 2 (R = Et) as the electrocatalyst, giving α-ketocarboxylates R′COCOO− and EtS− (Eq 7).69Methyl acrylate underwent similar reductive addition of CO2 under electrolysis conditions with 2.70
 | (6) |
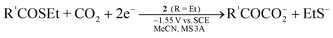 | (7) |
4. Nitrogenase-related transformations catalyzed by other cubane-type M4S4 clusters
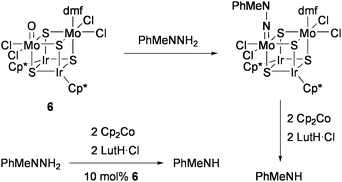
Several synthetic routes have been developed to prepare various heteronuclear cubane-type M4S4 clusters with metal compositions other than MFe3S4 (M = Fe, Mo, V). The catalytic activities of those clusters for nitrogenase-related transformations have been investigated by our group.The Mo2Ir2S4 cluster [(MoOCl2){MoCl2(dmf)}(IrCp*)2S4] (6: Cp* = η5-C5Me5) catalyzed the reduction of organohydrazine MePhNNH2 at room temperature in the presence of Cp2Co and LutH·Cl to give MePhNH.71 The Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety in 6 smoothly condensed with the NH2 group of MePhNNH2 to form the hydrazido(2−) ligand as the possible intermediate of the reductive N–N bond cleavage (Scheme 4). The isolated cluster [{Mo(NNMePh)Cl2}{MoCl2(dmf)}(IrCp*)2S4] liberated PhMeNH (21% yield) by treatment with Cp2Co and LutH·Cl (each two equiv). Replacement of the two Ir sites in 6 by Rh and/or of two of the sulfide ligands by selenides altered the catalytic activity to some extent (TON = 3–7).
O moiety in 6 smoothly condensed with the NH2 group of MePhNNH2 to form the hydrazido(2−) ligand as the possible intermediate of the reductive N–N bond cleavage (Scheme 4). The isolated cluster [{Mo(NNMePh)Cl2}{MoCl2(dmf)}(IrCp*)2S4] liberated PhMeNH (21% yield) by treatment with Cp2Co and LutH·Cl (each two equiv). Replacement of the two Ir sites in 6 by Rh and/or of two of the sulfide ligands by selenides altered the catalytic activity to some extent (TON = 3–7).
 | ||
| Scheme 4 Reduction of organohydrazine via formation of hydrazido(2−) ligand on the Mo2Ir2S4 cluster. | ||
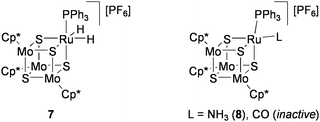
Disproportionation of hydrazine (Eq 2) was investigated by using the RuMo3S4 clusters in THF solution.72 In the reaction of [{RuH2(PPh3)}(MoCp*)3S4][PF6] (7: Chart 8) with 20 equiv of anhydrous hydrazine at 60 °C, 11.5 equiv of hydrazine was converted after 48 h to give ammonia and N2 in an almost 4:1 ratio. The conversion rate of hydrazine increased to 15.2 equiv after 18 h by changing the phosphine ligand for P(cyclohexyl)3. From the reaction mixture using 7, the ammonia cluster [{Ru(NH3)(PPh3)}(MoCp*)3S4][PF6] (8) was isolated, which exhibited a catalytic activity comparable to 7. In contrast, [{Ru(CO)(PPh3)}(MoCp*)3S4][PF6] was inactive, presumably because of the difficulty in replacing the CO ligand by hydrazine. Treatment of 7 with 20 equiv of phenylhydrazine at 60 °C for 18 h produced aniline (2.3 equiv), ammonia (1.2 equiv), N2 (1.6 equiv), and benzene (2.1 equiv), in addition to cluster 8.
 | ||
| Chart 8 RuMo3S4 clusters active for disproportionation of hydrazine. | ||
Recently, Mizobe and co-workers have synthesized the RuIr3S4 cluster binding molecular N2 at its Ru site, [{Ru(N2)(tmeda)}(IrCp*)3S4] (tmeda = Me2NCH2CH2NMe2: Chart 9).73 At present, this is the only example of the stable coordination of N2 to transition metal clusters containing bridging sulfide ligands. Although the N2 ligand was not transformed into ammonia, the N2cluster catalytically reduced proton to H2 by using various acids ([LutH][BF4], [Et3NH][BF4], etc.) and Cp2Co as the proton and the electron sources.74
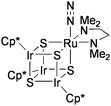 | ||
| Chart 9 The cubane-type cluster binding molecular N2. | ||
5. Organic transformations catalyzed by cubane-type M4S4 clusters
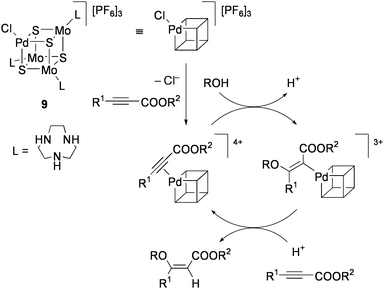
Hidai and co-workers have investigated the synthesis and reactivity of cubane-type M4S4 clusters containing noble metals in particular, which are not found in natural enzymes, because these clusters are expected to exhibit unique catalytic activities for organic synthesis.10The PdMo3S4 cluster [(PdCl){Mo(tacn)}3S4][PF6]3 (9: tacn = 1,4,7-triazacyclononane) was found to be a highly efficient catalyst for the stereoselective addition of alcohols to electron-deficient alkynes. Treatment of HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCOOMe with 0.06 mol% 9 in a MeOH solution at 40 °C for 24 h produced (Z)-MeOCHCHCOOMe in 95% yield.75 The product selectivity was so high as that concomitant formations of (E)-MeOCHCHCOOMe and (MeO)2CHCH2COOMe were inhibited to each less than 1%. This reaction was applicable to various combinations of terminal and internal alkynoic acid esters R1CCCOOR2 (R1 = H, Me, Ph, COOMe; R2 = alkyl, aryl) with alcohols ROH (R = Me, Et, CH2Ph), and the trans addition products (Z)-ROCR1CHCOOR2 were selectively obtained. Preservation of the cluster structure during the course of catalysis was confirmed by monitoring UV-vis spectra of the reaction solution.
CCOOMe with 0.06 mol% 9 in a MeOH solution at 40 °C for 24 h produced (Z)-MeOCHCHCOOMe in 95% yield.75 The product selectivity was so high as that concomitant formations of (E)-MeOCHCHCOOMe and (MeO)2CHCH2COOMe were inhibited to each less than 1%. This reaction was applicable to various combinations of terminal and internal alkynoic acid esters R1CCCOOR2 (R1 = H, Me, Ph, COOMe; R2 = alkyl, aryl) with alcohols ROH (R = Me, Et, CH2Ph), and the trans addition products (Z)-ROCR1CHCOOR2 were selectively obtained. Preservation of the cluster structure during the course of catalysis was confirmed by monitoring UV-vis spectra of the reaction solution.
A plausible mechanism for this catalysis is shown in Scheme 5. Initially, the alkynoic acid ester coordinates to the Pd site of the cluster to form the π-alkyne complex. Successive nucleophilic attack of alcohol to the electron-deficient carbon atom from the outer coordination sphere leads to the formation of the vinylpalladium intermediate. Protonolysis of the Pd–C bond with retention of the stereochemistry around the double bond affords the trans addition product. Although the π-alkyne intermediate can not be clearly observed, it is evidenced from the π-coordination of alkenes to the Pd site of 9 and the well characterized alkyne adduct of the related NiMo3S4 cluster (vide infra). It is noteworthy that conventional mononuclear Pd complexes do not work as effective catalysts in this reaction. The unique activity of 9 may originate from the tetrahedral Pd(II)-like site, which is surrounded by three firmly bonded sulfido ligands and provides only a single vacant site. The electron-poor nature of the Pd atom is deduced from the high CO stretching frequency of [{(Pd(CO)}{Mo(tacn)}3S4][Cl][PF6]3 (2085 cm−1). The Fenske-Hall molecular orbital calculations of the PdMo3S4 motif, where a zero oxidation state is assigned to the Pd atom, show that the Pd atom shares electron density through the formation of metal–metal bonds with the Mo(IV)3 triangle.76
Cluster
9 also catalyzed the stereoselective addition of carboxylic acids to electron-deficient alkynes. Reactions of HCCCOOMe with 3 equiv of acetic acid in an acetonitrile solution proceeded in the presence of catalytic amounts of 9 and NEt3 to produce MeCOOCHCHCOOMe with a stereoselectivity of Z/E = 98/2 (Eq 8).77 Various aliphatic and aromatic carboxylic acids as well as substituted acrylic acids added to HCCCOOMe stereoselectively. Other acetylenes with electron-withdrawing groups (including ester, amide, or sulfone) underwent Z-selective addition of acetic acid. Only the reaction between HCCCOPh and acetic acid exceptionally afforded the E-vinyl ketone probably due to the facile Z to Eisomerization of the product. A mechanism analogous to Scheme 5 has been proposed.
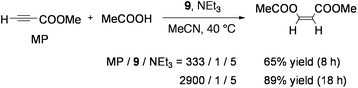 | (8) |
Intramolecular addition of acids smoothly takes place even to the non-activated alkyne moieties. Cyclization of alkynoic acids into enol lactones proceeded under mild conditions (20–40 °C, in acetonitrile) in the presence of 0.33 mol% 9 and 3.3 mol% NEt3 (Chart 10).78 Several complexes or salts of Rh, Pd, Ag, and Hg also catalyze these reactions, but the activities of these are much lower than 9. For example, catalytic rates observed for 9 were 12–17 times higher than the mononuclear complex [PdCl2(PhCN)2]. 4-Pentynoic acid and 5-hexynoic acid were selectively converted to γ- and δ-lactones, respectively. Catalytic rates for γ-lactonization were remarkably high, and the turnover number of the cyclization of 4-pentynoic acid reached 100000 after 19 h at 40 °C (300 times within 6 min at 20 °C). Reaction of an internal alkyne 4-hexynoic acid gave a mixture of γ- and δ-lactones, while only γ-lactone was formed from 3-decynoic acid. The cyclization of 6-heptynoic acid to the corresponding ε-lactone was very slow.
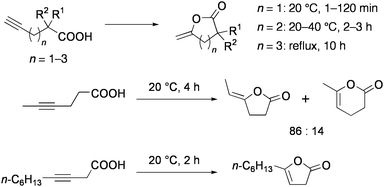 | ||
| Chart 10 Cyclization of alkynoic acids catalyzed by 9. | ||
Although less active than 9, the NiMo3S4 clusters [{Ni(L)}(MoCp*)3S4][PF6] (L = 1/2 1,5-cyclooctadiene (10a), dimethyl acetylenedicarboxylate (10b): Chart 11) also catalyze these lactone formations.79 The clusters that contain no noble metals work efficiently as catalysts under conditions similar to the above reactions with 9 (0.33 mol% NiMo3S4, 3.3 mol% NEt3, room temperature) in CH2Cl2 solution. The activity of 10 was much superior than [PdCl2(PhCN)2] in all cases, and the reaction was scarcely promoted by mononuclear Ni compounds or by the incomplete cubane-type cluster [(MoCp*)3S4][PF6]. It is notable that the π-coordination of alkyne to the Ni site has been crystallographically confirmed for the alkyne adduct 10b. This supports the validity of the catalytic mechanism shown in Scheme 5 for both 9 and 10, which have the same electronic configurations.
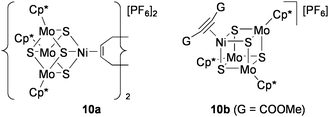 | ||
| Chart 11 NiMo3S4 clusters active for cyclization of alkynoic acids. | ||
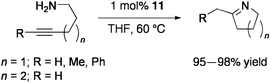
The related PdMo3S4 clusters [{Pd(L)}(Cp*Mo)3S4][PF6] (L = dibenzalacetone (11), maleic anhydride) catalyze cyclization of aminoalkynes to cyclic imines.80 In the presence of 1 mol% 11, 5-amino-1-phenyl-1-pentyne was quantitatively converted into 2-benzyl-1-pyrroline at 60 °C for 0.5 h in THF (Eq 9). Five- and six-membered cyclic imines were prepared in high yields.
 | (9) |
Recently, 11 was found to be effective for direct amination of allylic alcohols.81 When an equimolar mixture of allyl alcohol and secondary amine were refluxed in CH2Cl2 with 0.5 mol% 11 and 0.5 equiv of boric acid (or PhB(OH)2, B(OMe)3), N-allylamine was obtained in high yield. Only [Pd(PPh3)4] promotes such reactions among conventional Pd(0) and Pd(II) complexes, but this as well as 10a are less active than 11. Even amide was allylated by this method, while allylation of primary amines afforded some amounts of diallylamines. From the reactions of both α- and γ-monosubstituted allyl alcohols, only the γ-monosubstituted allyl amines were obtained. This suggests the catalytic cycle involving the formation of a π-allyl Pd complex as shown in Scheme 6, in which amines may attack the less hindered carbon atom of the π-allyl intermediate. This mechanism has certain resemblance to the IspH mechanism shown in Scheme 3. In fact, the π-allyl cluster [{Pd(η3-C3H5)}(MoCp*)3S4][PF6]2 (12) was produced by treatment of 11 with allyl chloride and AgPF6, and was fully characterized by X-ray crystallography.82 Further, 12 efficiently catalyzed the above allylamine formation from allyl alcohols without addition of boron reagents. Allylation of active methylene compounds was also facilitated by 12.
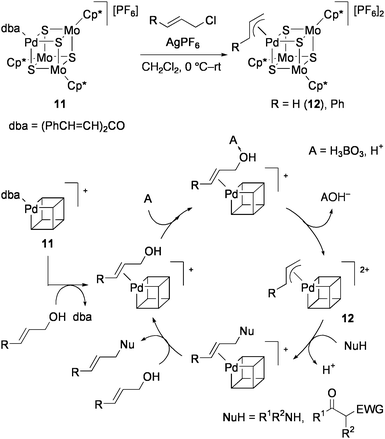 | ||
| Scheme 6 Proposed mechanism for the direct amination of allyl alcohol catalyzed by 11. | ||
Our preliminary studies have recently revealed that the cubane-type cluster [(RuCl3)(MoCp*)3S4]83 without any additives catalyzes the polymerization of methyl methacrylate (MMA).84 Thus, heating a solution of MMA (5.4 mL) in THF (17.5 mL) in the presence of the cubane-type cluster (10 mmol) at 80 °C for 6 h gave a polymer of MMA (1.25 g) with Mw = 360 000 and Mw/Mn = 2.9 (Mw = weight-averaged molecular weight, Mn = number-averaged molecular weight). Detailed investigations are now in progress to elucidate the mechanism of this polymerization, in comparison with the living radical polymerization of MMA by the combination of Ru(Ind)Cl(PPh3)2 (Ind = η5-C9H7) with an alkyl halide as an initiator.85
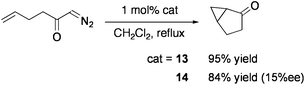
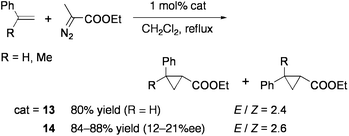
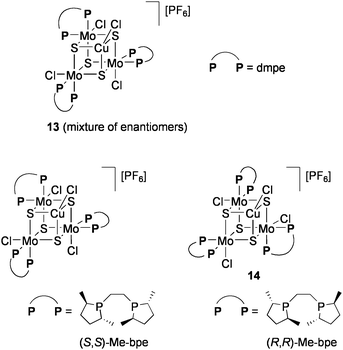
The groups of Llusar and Pérez-Prieto examined the addition reaction of carbenoids generated from diazo compounds by using the CuMo3S4 cluster [(CuCl){MoCl(dmpe)}3S4][PF6] (13: dmpe = Me2PCH2CH2PMe2: Chart 12).86 Intra- and intermolecular cyclopropanation of alkenes in the presence of 1 mol% 13 proceeded with high yields under reflux in CH2Cl2 (Eqs 10 and 11). The cluster remained intact after reaction. Optically active diphosphine ligands, 1,2-bis(2,5-dimethylphospholan-1-yl)ethane (Me-bpe), exert a chiral coordination environment on the CuMo3S4 core (Chart 12), and one of the enantiomers 14 have exhibited some asymmetric induction (up to 25% ee) in cyclopropanation. Based on the catalytic activities of Br- or Se-congeners of 14, the active site is considered to be generated by cleavage of one Cu–chalcogen bond.87
 | (10) |
 | (11) |
 | ||
| Chart 12 CuMoS4 catalysts for carbenoid addition. | ||
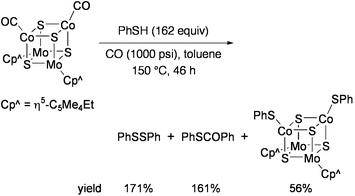
Sulfido clusters containing molybdenum and cobalt have been investigated in relevance to heterogeneous catalysts used for the industrial hydrodesulfurization (HDS) process.88 Curtis and co-workers studied the reactivities of Mo2Co2S3 and Mo2Co2S4 clusters toward desulfurization of organosulfur compounds. When [{Mo(η5-C5Me4Et)}2{Co(CO)}2S4] was heated with excess PhSH in toluene at 150 °C under CO pressure (1000 psi), PhSSPh and PhSCOPh were produced in addition to the cluster compound [{Mo(η5-C5Me4Et)}2{Co(SPh)}2S4] (Eq 12).89 Amount of PhSCOPh, which was produced via desulfurization of PhSH, was more than 100% per mole of the starting cluster.
 | (12) |
Several groups have prepared heterogeneous catalysts by incorporation of the cubane-type cluster [{Mo(H2O)3}3(NiCl)S4]3+ into zeolites by ion exchange90 or impregnation of the clusters [{Mo(η5-C5H4Me)}3(MLm)S4][p-MeC6H4SO3]n (M = Ru, Rh, Ir, Ni, Pd, Pt) on γ-alumina91 and employed the catalysts for CO hydrogenation, HDS, or hydrodenitrogenation reactions at high temperatures. However, since the structures of the catalytically active sites are not clear, the details are not described here.
Conclusion
Progress in catalysis by cubane-type M4S4 clusters has been summarized here. Intriguing catalysis by the Fe4S4 cluster will further be found in other metalloproteins, while synthetic M4S4 clusters would serve as unique catalytic precursors for organic synthesis that are not attainable by conventional metal complexes. It should be noted that only a few papers describing catalysis by synthetic M4S4 clusters have presented evidence to rule out the possibility that a small amount of active species formed via fragmentation of the cluster cores is responsible for the catalysis. However, the reactions generally proceed under mild conditions, so the relatively robust cubane-type structures are presumed to be fundamentally retained during the course of catalysis in most cases. We must await further investigations by both experimental and theoretical methods to elucidate the detailed reaction mechanisms and advantages of the polynuclear complexes.Acknowledgements
M.H. expresses his hearty thanks to Professor Yasushi Mizobe (1953–2010) for working together and collaborating with him for almost 30 years. H.S. expresses deep gratitude to Professor Y. Mizobe, who directed him to this research field, provided a fulfilling and comfortable environment for research, and had been giving warm advises and supports. His absence is a great loss to our family. The recent research by Y.M. and H.S. was supported by a Grant-in-Aid for Scientific Research (No. 18065005 on Priority Area “Chemistry of Concerto Catalysis” and No. 21350033 (B)) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- Reviews: R. Cammack, Adv. Inorg. Chem., 1992, 38 Search PubMed; H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 Search PubMed; M. K. Johnson, Curr. Opin. Chem. Biol., 1998, 2, 173–181 CrossRef CAS; J. B. Broderick, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 8, ch. 8.27, pp. 739–757 CrossRef CAS; M. S. Koay, M. L. Antonkine, W. Gärtner and W. Lubitz, Chem. Biodiversity, 2008, 5, 1571–1587 CrossRef CAS.
- B. R. Crane, L. M. Siegel and E. D. Getzoff, Science, 1995, 270, 59 CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS; Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567–572 CrossRef CAS.
- Fe–S clusters: R. H. Holm, Acc. Chem. Res., 1977, 10, 427–434 Search PubMed; A. Nakamura and N. Ueyama, Adv. Inorg. Chem., 1989, 33, 39–67 CrossRef CAS; P. Venkateswara Rao and R. H. Holm, Chem. Rev., 2004, 104, 527–559 CrossRef CAS.
- Fe–Mo–S clusters: D. Coucouvanis, Acc. Chem. Res., 1991, 24, 1–8 Search PubMed; S. M. Malinak and D. Coucouvanis, Prog. Inorg. Chem., 2001, 49, 599–685 CrossRef CAS.
- Fe–S and Fe–Mo–S clusters: R. A. Henderson, Chem. Rev., 2005, 105, 2365–2437 Search PubMed; R. A. Henderson, Coord. Chem. Rev., 2005, 249, 1841–1856 CrossRef.
- Non-Fe based clusters: S. Harris, Polyhedron, 1989, 8, 2843–2882 Search PubMed; T. Shibahara, Adv. Inorg. Chem., 1991, 37, 143–173 CrossRef CAS; T. Shibahara, Coord. Chem. Rev., 1993, 123, 73–147 CrossRef CAS; G. Sakane and T. Shibahara, in Transition Metal Sulfur Chemistry: Biological and Industrial Significance, ed. E. I. Stiefel and K. Matsumoto, American Chemical Sociey, Washington DC, 1996, ch. 13, pp. 225–239 CrossRef CAS; R. Hernandez-Molina and A. G. Sykes, Coord. Chem. Rev., 1999, 187, 291–302 CrossRef CAS; R. Hernandez-Molina and A. G. Sykes, J. Chem. Soc., Dalton Trans., 1999, 3137–3148 Search PubMed; R. Hernandez-Molina, M. N. Sokolov and A. G. Sykes, Acc. Chem. Res., 2001, 34, 223–230 CrossRef CAS.
- M. Hidai, S. Kuwata and Y. Mizobe, Acc. Chem. Res., 2000, 33, 46–52 CrossRef CAS; R. Llusar and S. Uriel, Eur. J. Inorg. Chem., 2003, 1271–1290 CrossRef CAS.
- M. Hidai and Y. Mizobe, in Transition Metal Sulfur Chemistry: Biological and Industrial Significance, ed. E. I. Stiefel and K. Matsumoto, American Chemical Society, Washington DC, 1996, ch. 19, pp. 310–323 Search PubMed; M. Hidai, in Perspectives in Organometallic Chemistry, ed. C. G. Screttas and B. R. Steele, The Royal Society of Chemistry, Special Publication No.287, Cambridge, 2003, pp. 62–73 Search PubMed; Y. Ishii and M. Hidai, in Multimetallic Catalysts in Organic Synthesis, ed. M. Shibasaki and Y. Yamamoto, Wiley-VHC, Weinheim, 2004, ch. 9, pp. 201–223 Search PubMed; M. Hidai and Y. Mizobe, Can. J. Chem., 2005, 83, 358–374 Search PubMed; Y. Mizobe, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova, The Royal Society of Chemistry, Cambridge, 2007, ch. 11.2, pp. 714–730 Search PubMed.
- Although most of the cubane-type clusters described here have metal–metal bonds in the cluster core, these bonds are omitted for clarity from illustrations of molecular structures throughout this report.
- Reviews: P. A. Frey and O. Th. Magnusson, Chem. Rev., 2003, 103, 2129–2148 Search PubMed; P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS; A. Marquet, B. T. S. Bui, A. G. Smith and M. J. Warren, Nat. Prod. Rep., 2007, 24, 1027–1040 CrossRef CAS; K. S. Duschene, S. E. Veneziano, S. C. Silver and J. B. Broderick, Curr. Opin. Chem. Biol., 2009, 13, 74–83 RSC.
- Reviews: H. Beinert, M. C. Kennedy and C. D. Stout, Chem. Rev., 1996, 96, 2335–2373 Search PubMed; D. H. Flint and R. M. Allen, Chem. Rev., 1996, 96, 2315–2334 CrossRef CAS.
- A. H. Robbins and C. D. Stout, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3639–3643 CAS; H. Lauble, M. C. Kennedy, H. Beinert and C. D. Stout, Biochemistry, 1992, 31, 2735–2748 CrossRef CAS; H. Lauble, M. C. Kennedy, M. H. Emptage, H. Beinert and C. D. Stout, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13699–13703 CrossRef CAS.
- T. Gräwert, I. Span, W. Eisenreich, F. Rohdich, J. Eppinger, A. Bacher and M. Groll, Proc. Natl. Acad. Sci., 2010, 107, 1077–1081 CrossRef.
- W. Wang, K. Wang, Y.-L. Liu, J.-H. No, J. Li, M. J. Nilges and E. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4522–4527 CrossRef CAS; K. Wang, W. Wang, J.-H. No, Y. Zhang, Y. Zhang and E. Oldfield, J. Am. Chem. Soc., 2010, 132, 6719–6727 CrossRef CAS.
- W. Wang, J. Li, K. Wang, C. Huang, Y. Zhang and E. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11189–11193 CrossRef CAS.
- W. Xu, N. S. Lees, D. Adedeji, J. Wiesner, H. Jomaa, B. M. Hoffman and E. C. Duin, J. Am. Chem. Soc., 2010, 132, 14509–14520 CrossRef CAS.
- T. Herskovitz, B. A. Averill, R. H. Holm, J. A. Ibers, W. D. Phillips and J. F. Weiher, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2437–2441 CrossRef CAS.
- C. J. A. Daley and R. H. Holm, Inorg. Chem., 2001, 40, 2785–2793 CrossRef CAS.
- C. J. A. Daley and R. H. Holm, J. Inorg. Biochem., 2003, 97, 287–298 CrossRef CAS.
- T. D. P. Stack and R. H. Holm, J. Am. Chem. Soc., 1988, 110, 2484–2494 CrossRef CAS; J. A. Weigel and R. H. Holm, J. Am. Chem. Soc., 1991, 113, 4184–4191 CrossRef CAS; L. Cai and R. H. Holm, J. Am. Chem. Soc., 1994, 116, 7177–7188 CrossRef CAS; P. V. Rao and R. H. Holm, Chem. Rev., 2004, 104, 527–559 CrossRef CAS , and references therein.
- A. K. Yatsimirsky and A. I. Diky, Inorg. Chim. Acta, 1991, 186, 161–163 CrossRef.
- M. Tezuka, T. Yajima, A. Tsuchiya, Y. Matsumoto, Y. Uchida and M. Hidai, J. Am. Chem. Soc., 1982, 104, 6834–6836 CrossRef CAS; M. Nakazawa, Y. Mizobe, Y. Matsumoto, Y. Uchida, M. Tezuka and M. Hidai, Bull. Chem. Soc. Jpn., 1986, 59, 809–814 CAS.
- T. Tomohiro, K. Uoto and H. Okuno, J. Chem. Soc., Chem. Commun., 1990, 194–195 RSC.
- T. Nagano, K. Yoshikawa and M. Hirobe, Tetrahedron Lett., 1980, 21, 297–300 CrossRef CAS; I. Tabushi, Y. Kuroda and Y. Sasaki, J. Mol. Catal., 1988, 49, 1–7 CrossRef CAS.
- T. Ito, T. Nagano and M. Hirobe, Chem Pharm. Bull., 1986, 34, 2013–2017 CAS.
- H. Inoue, M. Shirai and E. Haruki, J. Chem. Soc., Chem. Commun., 1987, 674–675 RSC.
- N. Ueyama, T. Sugawara, A. Kajiwara and A. Nakamura, J. Chem. Soc., Chem. Commun., 1986, 434–435 RSC; T. Sugawara, N. Ueyama and A. Nakamura, J. Chem. Soc., Dalton Trans., 1991, 249–253 RSC.
- N. Ueyama, T. Sugawara, A. Nakamura, K. Yamaguchi and T. Fueno, Chem. Lett., 1988, 223–224 CrossRef CAS.
- A. Nakamura, M. Kamada, Sugihashi and S. Otsuka, J. Mol. Catal., 1980, 8, 353–367 CrossRef CAS.
- T. Ito, T. Nagano and M. Hirobe, Tetrahedron Lett., 1980, 21, 1343–1246 CrossRef.
- T. Ito, T. Nagano, M. Sato and M. Hirobe, Tetrahedron Lett., 1989, 30, 6387–6388 CrossRef CAS.
- R. S. McMillan, J. Renaud, J. G. Reynolds and R. H. Holm, J. Inorg. Biochem., 1979, 11, 213–227 CrossRef CAS.
- K. L. C. Grönberg, R. A. Henderson and K. E. Oglieve, J. Chem. Soc., Dalton Trans., 1998, 3093–3104 RSC.
- K. Tanaka, M. Nakamoto, M. Tsunomori and T. Tanaka, Chem. Lett., 1987, 613–616 CAS.
- H. Inoue and M. Suzuki, J. Chem. Soc., Chem. Commun., 1980, 817–818 RSC; H. Inoue and M. Sato, J. Chem. Soc., Chem. Commun., 1983, 983–984 RSC.
- J. Kim and D. C. Rees, Science, 1992, 257, 1677–1682 CrossRef CAS; O. Einsle, F. A. Tezcan, S. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS.
- S. P. Cramer, K. O. Hodgson, W. O. Gillum and L. E. Mortenson, J. Am. Chem. Soc., 1978, 100, 3398–3407 CrossRef CAS.
- J. M. Arber, B. R. Dobson, R. R. Eady, P. Stevens, S. S. Hasnain, C. D. Garner and B. E. Smith, Nature, 1987, 325, 372 CrossRef CAS.
- G. N. Goerge, C. L. Coyle, B. J. Hales and S. P. Cramer, J. Am. Chem. Soc., 1988, 110, 4057–4059 CrossRef CAS.
- G. Christou, C. D. Garner and F. E. Mabbs, J. Chem. Soc., Chem. Commun., 1978, 740–741 RSC; G. Christou, C. D. Garner and F. E. Mabbs, Inorg. Chim. Acta, 1978, 28, L189–L190 CrossRef.
- T. E. Wolff, J. M. Berg, C. Warrick, K. O. Hodgson, R. H. Holm and R. B. Frankel, J. Am. Chem. Soc., 1978, 100, 4630–4632 CrossRef CAS; T. E. Wolff, J. M. Berg, K. O. Hodgson, R. B. Frankel and R. H. Holm, J. Am. Chem. Soc., 1979, 101, 4140–4150 CrossRef CAS.
- W. H. Armstrong, P. K. Mascharak and R. H. Holm, Inorg. Chem., 1982, 21, 1699–1701 CrossRef CAS; W. H. Armstrong, P. K. Mascharak and R. H. Holm, J. Am. Chem. Soc., 1982, 104, 4373–4383 CrossRef CAS.
- R. E. Palermo, R. Singh, J. K. Bashkin and R. H. Holm, J. Am. Chem. Soc., 1984, 106, 2600–2612 CrossRef CAS.
- T. Yamamura, G. Christou and R. H. Holm, Inorg. Chem., 1983, 22, 939–949 CrossRef CAS; Y. Mizobe, P. K. Mascharak, R. E. Palermo and R. H. Holm, Inorg. Chim. Acta, 1983, 80, L65–L67 CrossRef CAS.
- R. E. Palermo and R. H. Holm, J. Am. Chem. Soc., 1983, 105, 4310–4318 CrossRef CAS.
- D. Coucouvanis, K. D. Demadis, S. M. Malinak, P. E. Mosier, M. A. Tyson and L. J. Laughlin, J. Mol. Catal. A: Chem., 1996, 107, 123–135 CrossRef CAS.
- D. Coucouvanis, P. E. Mosier, K. D. Demadis, S. Patton, S. M. Malinak, C. G. Kim and M. A. Tyson, J. Am. Chem. Soc., 1993, 115, 12193–12194 CrossRef CAS.
- K. D. Demadis, S. M. Malinak and D. Coucouvanis, Inorg. Chem., 1996, 35, 4038–4046 CrossRef CAS.
- P. R. Challen, S.-M. Koo, C. G. Kim, W. R. Dunham and D. Coucouvanis, J. Am. Chem. Soc., 1990, 112, 8606–8607 CrossRef CAS; P. E. Mosier, C. G. Kim and D. Coucouvanis, Inorg. Chem., 1993, 32, 2620–2621 CrossRef CAS.
- K. D. Demadis and D. Coucouvanis, Inorg. Chem., 1994, 33, 4195–4197 CrossRef CAS; K. D. Demadis and D. Coucouvanis, Inorg. Chem., 1995, 34, 3658–3666 CrossRef CAS.
- L. J. Laughlin and D. Coucouvanis, J. Am. Chem. Soc., 1995, 117, 3118–3125 CrossRef CAS.
- S. M. Malinak, A. M. Simeonov, P. E. Mosier, C. E. McKenna and D. Coucouvanis, J. Am. Chem. Soc., 1997, 119, 1662–1667 CrossRef CAS.
- J. A. Kovacs and R. H. Holm, J. Am. Chem. Soc., 1986, 108, 340–341 CrossRef CAS; J. A. Kovacs and R. H. Holm, Inorg. Chem., 1987, 26, 702–711 CrossRef CAS.
- S. M. Malinak, K. D. Demadis and D. Coucouvanis, J. Am. Chem. Soc., 1995, 117, 3126–3133 CrossRef CAS.
- K. Tanaka, M. Moriya and T. Tanaka, Chem. Lett., 1987, 373–376 CrossRef CAS; K. Tanaka, M. Moriya, S. Uezumi and T. Tanaka, Inorg. Chem., 1988, 27, 137–143 CrossRef CAS.
- Y. Hozumi, Y. Imasaka, K. Tanaka and T. Tanaka, Chem. Lett., 1983, 897–900 CAS.
- K. Tanaka, M. Tanaka and T. Tanaka, Chem. Lett., 1981, 895–898 CrossRef CAS . The catalyst used in this report is probably 2a despite description as [Mo2Fe6S9(SPh)8]3−.
- K. Tanaka, M. Honjo and T. Tanaka, J. Inorg. Biochem., 1984, 22, 187–199 CrossRef CAS; K. Tanaka, M. Nakamoto, Y. Tashiro and T. Tanaka, Bull. Chem. Soc. Jpn., 1985, 58, 316–321 CrossRef CAS.
- S. Kuwabata, K. Tanak and T. Tanaka, Inorg. Chem., 1986, 25, 1691–1697 CrossRef CAS.
- K. Tanaka, Y. Imasaka, M. Tanaka, M. Honjo and T. Tanaka, J. Am. Chem. Soc., 1982, 104, 4258–4260 CrossRef CAS.
- S. Kuwabata, Y. Hozumi, K. Tanaka and T. Tanaka, Chem. Lett., 1985, 401–404 CrossRef CAS.
- Y. Imasaka, K. Tanaka and T. Tanaka, Chem. Lett., 1983, 1477–1480 CrossRef CAS.
- K. Tanaka, S. Kuwabata, S. Denno and T. Tanaka, Bull. Chem. Soc. Jpn., 1989, 62, 1561–1566 CrossRef CAS.
- K. Tanaka, S. Uezumi and T. Tanaka, J. Chem. Soc., Dalton Trans., 1989, 1547–1554 RSC.
- S. Kuwabata, S. Uezumu, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1986, 135–136 RSC; S. Kuwabata, S. Uezumi, K. Tanaka and T. Tanaka, Inorg. Chem., 1986, 25, 3018–3022 CrossRef CAS; K. Tanaka, T. Matsui and T. Tanaka, Chem. Lett., 1989, 1827–1830 CrossRef CAS; K. Tanaka, N. Komeda and T. Matsui, Inorg. Chem., 1991, 30, 3282–3288 CrossRef CAS.
- K. Tanaka, R. Wakita and T. Tanaka, Chem. Lett., 1987, 1951–1954 CAS; K. Tanaka, R. Wakita and T. Tanaka, J. Am. Chem. Soc., 1989, 111, 2428–2433 CrossRef CAS.
- K. Tanaka, T. Matsui and T. Tanaka, J. Am. Chem. Soc., 1989, 111, 3765–3767 CrossRef CAS; N. Komeda, H. Nagao, T. Matsui, G. Adachi and K. Tanaka, J. Am. Chem. Soc., 1992, 114, 3625–3630 CrossRef CAS.
- H. Nagao, H. Miyamoto and K. Tanaka, Chem. Lett., 1991, 323–326 CAS.
- T. Masumori, H. Seino, Y. Mizobe and M. Hidai, Inorg. Chem., 2000, 39, 5002–5003 CrossRef CAS; H. Seino, T. Masumori, M. Hidai and Y. Mizobe, Organometallics, 2003, 22, 3424–3431 CrossRef CAS.
- I. Takei, K. Dohki, K. Kobayashi, T. Suzuki and M. Hidai, Inorg. Chem., 2005, 44, 3768–3770 CrossRef.
- H. Mori, H. Seino, M. Hidai and Y. Mizobe, Angew. Chem., Int. Ed., 2007, 46, 5431–5434 CrossRef CAS.
- H. Seino, H. Mori, K. Hirata and Y. Mizobe, unpublished results.
- T. Murata, Y. Mizobe, H. Gao, Y. Ishii, T. Wakabayashi, F. Nakano, T. Tanase, S. Yano, M. Hidai, I. Echizen, H. Nanikawa and S. Motomura, J. Am. Chem. Soc., 1994, 116, 3389–3398 CrossRef CAS.
- C. S. Bahn, A. Tan and S. Harris, Inorg. Chem., 1998, 37, 2770–2778 CrossRef CAS.
- T. Wakabayashi, Y. Ishii, T. Murata, Y. Mizobe and M. Hidai, Tetrahedron Lett., 1995, 36, 5585–5588 CrossRef CAS.
- T. Wakabayashi, Y. Ishii, K. Ishikawa and M. Hidai, Angew. Chem., Int. Ed. Engl., 1996, 35, 2123–2124 CrossRef CAS.
- I. Takei, Y. Wakebe, K. Suzuki, Y. Enta, T. Suzuki, Y. Mizobe and M. Hidai, Organometallics, 2003, 22, 4639–4641 CrossRef CAS.
- I. Takei, Y. Enta, Y. Wakebe, T. Suzuki and M. Hidai, Chem. Lett., 2006, 35, 590–4591 CrossRef CAS.
- Y. Tao, Y. Zhou, J. Qu and M. Hidai, Tetrahedron Lett., 2010, 51, 1982–1984 CrossRef CAS.
- Y. Tao, B. Wang, B. Wang, L. Qu and J. Qu, Org. Lett., 2010, 12, 2726–2729 CrossRef CAS.
- I. Takei, K. Kobayashi, K. Dohki, S. Nagao, Y. Mizobe and M. Hidai, Chem. Lett., 2007, 36, 546–547 CrossRef CAS.
- M. Nabika and M. Hidai, unpublished results.
- H. Takahashi, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 3820–3823 CrossRef CAS.
- M. Feliz, E. Guillamón, R. Llusar, C. Vicent, S.-E. Stiriba, J. Pérez-Prieto and M. Barberis, Chem.–Eur. J., 2006, 12, 1486–1492 CrossRef CAS.
- E. Guillamón, R. Llusar, J. Pérez-Prieto and S.-E. Stiriba, J. Organomet. Chem., 2008, 693, 1723–1727 CrossRef CAS.
- M. D. Curtis, in Transition Metal Sulfur Chemistry: Biological and Industrial Significance, ed. E. I. Stiefel and K. Matsumoto, American Chemical Sociey, Washington DC, 1996, ch. 8, pp. 154–175 Search PubMed.
- M. A. Mansour, M. D. Curtis and J. W. Kampf, Organometallics, 1995, 14, 5460–5462 CrossRef CAS ; 1997, 16, 3363–3370.
- M. Taniguchi, Y. Ishii, T. Murata, T. Tatsumi and M. Hidai, J. Chem. Soc., Chem. Commun., 1995, 2533–2534 RSC; T. Tatsumi, M. Taniguchi, S. Yasuda, Y. Ishii, T. Murata and M. Hidai, Appl. Catal., A, 1996, 139, L5–L10 CrossRef CAS; M. Taniguchi, D. Imamura, H. Ishige, Y. Ishii, T. Murata, M. Hidai and T. Tatsumi, J. Catal., 1999, 187, 139–150 CrossRef CAS.
- K. Herbst, M. Brorson and A. Carlsson, J. Mol. Catal. A: Chem., 2010, 325, 1–7 CrossRef CAS.
Footnote |
| † In memory of Professor Yasushi Mizobe |
| This journal is © The Royal Society of Chemistry 2011 |