A synthesis of strychnine by a longest linear sequence of six steps†
David B. C.
Martin
and
Christopher D.
Vanderwal
*
Department of Chemistry, University of California, 1102 Natural Sciences II Mail Code: 2025, Irvine, California CA 92697, USA. E-mail: cdv@uci.edu
First published on 4th February 2011
Abstract
Strychnine is synthesized via a longest linear sequence of six steps from commercially available starting materials. Key steps include a base-mediated intramolecular Diels–Alder reaction of a tryptamine-derived Zincke aldehyde, a Ru-catalyzed trans-hydrosilylation of 1,4-butynediol, and a tandem Brook rearrangement/intramolecular conjugate addition reaction that affords the Wieland–Gumlich aldehyde.
Introduction
Known for centuries as a powerful toxin and as the flagship member of the Strychnosalkaloids, strychnine (1, Scheme 1) can be considered a benchmark for the state-of-the art in alkaloid synthesis strategy.1,2 Beginning with Woodward's inaugural achievement2a and in the course of more than a dozen instructive syntheses that followed, this alkaloid target has elicited many different strategies to its complex heptacyclic architecture, with attendant advances in methodology in many cases. The structure of the Wieland–Gumlich aldehyde (2),3 itself a natural product (also named caracurine VII) and the penultimate intermediate in many previous syntheses of strychnine,2b,c,f,g,l–o,4 stimulated our group's interest in developing the intramolecular cycloaddition reactions of tryptamine-derived Zincke aldehydes, a strategy that we recently deployed in a short synthesis of the relatively simple Strychnosalkaloid norfluorocurarine.5,6 In this communication, we disclose how application of this general strategy led to the shortest synthesis of strychnine to date—commercially available, inexpensive compounds are converted into the classic target via a longest linear sequence of only six steps.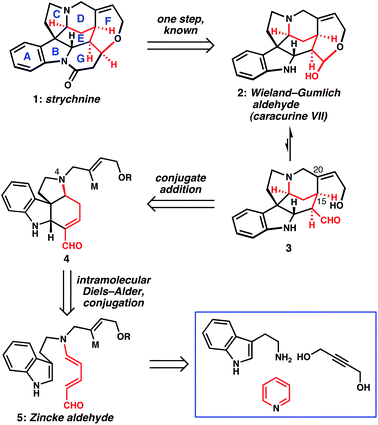 | ||
| Scheme 1 Retrosynthetic analysis of strychnine demonstrates the powerfully simplifying Zincke aldehyde cycloaddition disconnection (M = metal or suitable metal precursor). | ||
Synthesis plan
Although two previous approaches to strychnine have taken advantage of Diels–Alder cycloaddition reactions with the indole double bond serving as the 2π-component,2k,n our use of tryptamine-derived Zincke aldehydes in such reactions ideally positions appropriate functional groups with the correct oxidation state for an exceedingly short synthesis. Our strategy is illustrated in Scheme 1 in the form of a retrosynthetic analysis, wherein the hydroxy–aldehyde tautomer (3) of the Wieland–Gumlich aldehyde is first simplified by conceptual cleavage of the C15–C20 bond of the D-ring piperidine. In the forward sense, a C20 vinyl anion equivalent would engage an α,β-unsaturated aldehyde in a conjugate addition reaction. Tetracycle 4 would arise from tryptamine-derived Zincke aldehyde 5via our previously described base-mediated Diels–Alder reaction, which results in conjugation of the enal. The cycloaddition substrate would be accessed according to the Zincke process for the ring-opening of pyridinium salts.5,7 The nature of the N4 side chain would dictate the possibilities for D-ring closure, and we speculated that flexibility in this regard would be beneficial to reducing this plan to practice.Results and discussion
We sought substrates of type 5 that were competent in our formal cycloaddition and that bore a suitable group (M) to enable the formation of the D ring by a conjugate addition to the ring E enal in 4. In our previous application of the key base-mediated bicyclization reaction, we found that vinyl halides were susceptible to decompositionvia elimination pathways under the harsh reaction conditions (KOt-Bu, THF, 80 °C).5 Further investigations demonstrated that alkynes, free hydroxyl groups and several other useful functional groups were not tolerated by this reaction. These observations inspired us to consider tetracycle 7 (Scheme 2), bearing a secondary amine, as a key intermediate that could be N-alkylated to introduce desired functionality. Because of the limited stability of Zincke aldehydes derived from primary amines, our cycloaddition reaction could not be used to directly generate 7; rather, a suitable N-protected tetracyclic precursor (6) was needed. Requirements of the masking group were: 1. It could not be electron-deficient, because formation of Zincke aldehydes is only efficient for relatively electron-rich secondary amines; 2. It must be stable to the strongly basic conditions of the cycloaddition reaction; 3. Removal of the group must not be dependent upon reductive8 or acidic9 conditions. The following groups were stable to the conditions for cycloaddition: Me, Bn, PMB, 2,4-DMB, TMSCH2CH2, and allyl. After substantial experimentation with each of these cycloadducts, only the N-allyl group could be removed. Under palladium catalysis with excess N,N′-dimethylbarbituric acid as an allyl scavenger,10 the clean deallylation reaction was accompanied by Knoevenagel condensation of the barbituric acid derivative with the aldehyde followed by Michael addition of a second equivalent of nucleophile to afford adduct 8 in good yield. This undesired reactivity could be circumvented using a C-alkylated barbituric acid derivative, which precludes the dehydrative step of the Knoevenagel condensation; however, it was more convenient to use commercially available methyl Meldrum's acid, which also enabled clean deallylation to afford 7 (see below). Not surprisingly, the resultant product, which bears two nucleophilic nitrogen atoms along with two electrophilic carbons, was poorly stable. As a consequence, isolated yields of 7 were low, and storage led to degradation; however, in situ realkylation of the liberated secondary amine provided an attractive solution to this problem (see below).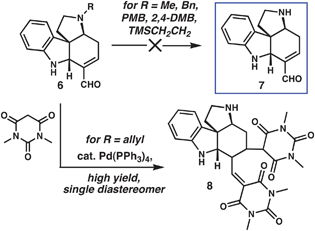 | ||
| Scheme 2 Attempts to access versatile NH tetracycle 7. | ||
With access to versatile yet unstable intermediate 7, we had flexibility to evaluate different N4 side chains and their reactivity in efforts to forge the C15–C20 bond of the D-ring piperidine. The approach described in Scheme 3 proved optimally concise. N-Allyl tryptamine 911 was converted to the Zincke aldehyde under standard conditions,5,6b–d and the base-mediated bicyclization reaction afforded 12. Previous routes to electrophiles related to 15 typically required upward of half a dozen steps; ultimately, these operations never served to lengthen the linear reaction sequence, because the synthesis of the core required more steps. Faced with the desire to maximize convergency and minimize step count, we were forced to advance a new and more direct solution. We recognized that the ruthenium-catalyzed trans-hydrosilylation of alkynes reported by Trost and Ball12 could serve as a pivotal reaction in the conversion of inexpensive 1,4-butynediol into a suitable N4 side chain precursor. Indeed, siloxacycle 14 was formed in high yield via this procedure and, with one of the two hydroxyl groups internally protected, the other was converted to the bromide under standard conditions. Methylmagnesium bromide served to open the siloxacycle, thereby generating the TMS group and liberating the hydroxyl group of 15. This sequence could be reliably carried out on gram-scale, with three-step yields reproducibly above 45%, and represents an efficient approach to such a polyfunctional, stereodefined trisubstituted alkene. When 15 was added to the reaction mixture after complete deallylation of 12 using methyl Meldrum's acid, refunctionalized core 16 was isolated in 69% yield. The free alcohol present in 16 enabled a Brook rearrangement13 of the corresponding alkoxide with presumed transmetalation to copper, followed by intramolecular conjugate addition to afford the Wieland–Gumlich aldehyde (2) in up to 10% isolated yield. This penultimate intermediate was converted to strychninevia the protocol of Robinson and Anet.2c,4
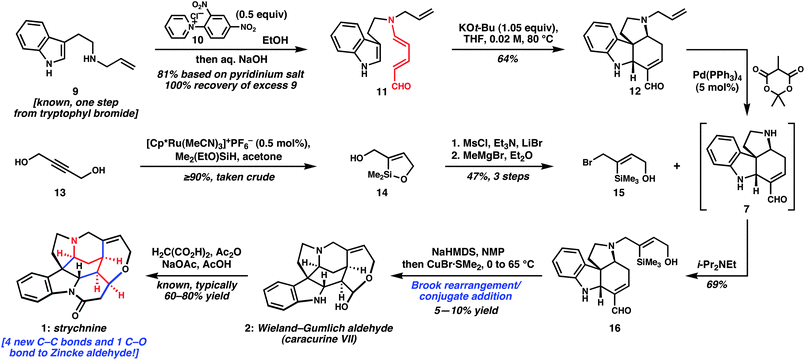 | ||
| Scheme 3 Synthesis of strychninevia a sequence of six linear steps. | ||
A brief discussion of the D-ring-forming step is in order.14,15 This specific type of Brook rearrangement finds precedent in the work of Takeda16 and more recently in the anion-relay chemistry of Smith.17 While these workers report many examples of high-yielding C–C bond formations after the initial rearrangement, our substantial efforts in optimization18 led to the best case as shown in Scheme 3. In most reactions, the mass balance consists largely of the product of protodesilylation, which likely occurs by Brook rearrangement/hydrolysis of the resulting vinyl metal. We believe that the conversion of 16 to 2 is self-limiting in the sense that the presence of acidic protons in the substrate and/or product leads to quenching of the key reactive organometallic reagent, resulting in an inefficient and rather irreproducible reaction. Nonetheless, adoption of this Brook rearrangement-based strategy enabled the completion of the synthesis in only six linear steps because of the rapid assembly of vinylsilane 15. Strategies to improve the overall yield at the expense of a slightly longer sequence are being assessed.
Conclusions
Our concise synthesis of strychnine clearly documents the utility of Zincke aldehydes for complex molecule synthesis—in the course of only four chemical steps, four new carbon–carbon bonds and one carbon–oxygen bond are forged to the five carbons of the donor–acceptor diene. Although the overall yield is not as high as desired, we argue that our strategy demonstrates excellent efficiency by the important metric of step economy.19 In addition to the concise synthesis of the classic target strychnine, this paper offers four-step access to tetracycle 7 or N-functionalized variants thereof, which should prove broadly useful for the synthesis of many other indole monoterpene alkaloids.Acknowledgements
We thank the NSF (CAREER Award CHE-0847061) for support of our work. D. B. C. M. is the recipient of graduate fellowships from Eli Lilly, the NSERC of Canada, and Bristol-Myers Squibb. C. D. V. is grateful for support through an Amgen Young Investigator Award and an AstraZeneca Award for Excellence in Chemistry. C. D. V. is a fellow of the A. P. Sloan Foundation. We thank Dr Martin Schnermann for helpful discussions.Notes and references
- For recent reviews, see: (a) J. Bonjoch and D. Solé, Chem. Rev., 2000, 100, 3455–3482 CrossRef CAS; (b) M. Mori, Heterocycles, 2010, 81, 259–292 CrossRef CAS.
- (a) R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, J. Am. Chem. Soc., 1954, 76, 4749–4751 CrossRef CAS; (b) P. Magnus, M. Giles, R. Bonnert, C. S. Kim, L. McQuire, A. Merritt and N. Vicker, J. Am. Chem. Soc., 1992, 114, 4403–4405 CrossRef CAS; (c) S. D. Knight, L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1993, 115, 9293–9294 CrossRef CAS; (d) M. E. Kuehne and F. Xu, J. Org. Chem., 1993, 58, 7490–7497 CrossRef CAS; (e) V. H. Rawal and S. Iwasa, J. Org. Chem., 1994, 59, 2685–2686 CrossRef CAS; (f) M. E. Kuehne and F. Xu, J. Org. Chem., 1998, 63, 9427–9433 CrossRef CAS; (g) D. Solé, J. Bonjoch, S. García-Rubio, E. Peidró and J. Bosch, Angew. Chem., Int. Ed., 1999, 38, 395–397 CrossRef CAS; (h) M. Ito, C. W. Clark, M. Mortimore, J. B. Goh and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 8003–8010 CrossRef CAS; (i) M. J. Eichberg, R. L. Dorta, K. Lamottke and K. P. C. Vollhardt, Org. Lett., 2000, 2, 2479–2481 CrossRef CAS; (j) M. Nakanishi and M. Mori, Angew. Chem., Int. Ed., 2002, 41, 1934–1936 CrossRef CAS; (k) G. J. Bodwell and J. Li, Angew. Chem., Int. Ed., 2002, 41, 3261–3262 CrossRef CAS; (l) T. Ohshima, Y. Xu, R. Takita, S. Shimuzu, D. Zhong and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14546–14547 CrossRef CAS; (m) Y. Kaburagi, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2004, 126, 10246–10247 CrossRef CAS; (n) H. Zhang, J. Boonsombat and A. Padwa, Org. Lett., 2007, 9, 279–282 CrossRef CAS; (o) G. Sirasani, T. Paul, W. Dougherty, Jr., S. Kassel and R. B. Andrade, J. Org. Chem., 2010, 75, 3529–3532 CrossRef CAS; (p) C. Beemelmanns and H.-U. Reissig, Angew. Chem., Int. Ed., 2010, 49, 8021–8025 CrossRef CAS.
- For a lead reference that includes the synthesis of 2 from strychnine, see: F. A. L. Anet and R. Robinson, J. Chem. Soc., 1955, 2253–2262 Search PubMed.
- For the conversion of the Wieland–Gumlich aldehyde to strychnine, see: F. A. L. Anet and R. Robinson, Chem. Ind., 1953, 245 Search PubMed.
- D. B. C. Martin and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 3472–3473 CrossRef CAS.
- For our previous work with Zincke aldehydes, see: (a) A. M. Kearney and C. D. Vanderwal, Angew. Chem., Int. Ed., 2006, 45, 7803–7806 CrossRef CAS; (b) S. E. Steinhardt, J. S. Silverston and C. D. Vanderwal, J. Am. Chem. Soc., 2008, 130, 7560–7561 CrossRef CAS; (c) T. D. Michels, J. U. Rhee and C. D. Vanderwal, Org. Lett., 2008, 10, 4787–4790 CrossRef CAS; (d) S. E. Steinhardt and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 7546–7547 CrossRef CAS; (e) T. D. Michels, M. J. Kier, A. M. Kearney and C. D. Vanderwal, Org. Lett., 2010, 12, 3093–3095 CrossRef.
- (a) T. Zincke, Justus Liebigs Ann. Chem., 1903, 330, 361–374 CAS; (b) T. Zincke, Justus Liebigs Ann. Chem., 1904, 333, 296–345 CrossRef; (c) T. Zincke and W. Wurker, Justus Liebigs Ann. Chem., 1905, 338, 107–141; (d) W. König, J. Prakt. Chem., 1904, 69, 105–137 CrossRef.
- The unsaturated aldehyde in 6 is easily reduced under many conditions.
- Under even relatively mildly acidic conditions, intermolecular condensation reactions between the aniline nitrogen and the aldehyde occur readily.
- F. Garro-Helion, A. Merzouk and F. Guibé, J. Org. Chem., 1993, 58, 6109–6113 CrossRef CAS.
- N-Allyl tryptamine is known, and is available in quantitative yield by treatment of tryptophyl bromide with an excess of allylamine; see the Supporting Information.† For a related preparation, see: S. F. Martin, S. A. Williamson, R. P. Gist and K. M. Smith, J. Org. Chem., 1983, 48, 5170–5180 Search PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS.
- A. G. Brook, Acc. Chem. Res., 1974, 7, 77–84 CrossRef CAS.
- Arguably, the best successes in the formation of the D ring via C15–C20 bond construction have involved Heck reactions, as pioneered by Rawal. See: (a) V. H. Rawal and C. Michoud, Tetrahedron Lett., 1991, 32, 1695–1698 CrossRef CAS; (b) V. H. Rawal, C. Michoud and R. F. Monestel, J. Am. Chem. Soc., 1993, 115, 3030–3031 CrossRef CAS; (c) ref. 2e .
- For a relevant discussion of the difficulty of C15–C20 bond construction, see ref. 2i.
- H. Taguchi, K. Ghoroku, M. Tadaki, A. Tsubouchi and T. Takeda, J. Org. Chem., 2002, 67, 8450–8456 CrossRef CAS.
- A. B. Smith III, W.-S. Kim and R. Tong, Org. Lett., 2010, 12, 588–591 CrossRef.
- In the course of close to 100 different experiments to ameliorate this reaction, we varied the Cu(I) salt, ligand for Cu, solvent, concentration, temperature, base, and additives, without success.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00009h |
| This journal is © The Royal Society of Chemistry 2011 |