Tuning and optimizing the intrinsic interactions between phthalocyanine-based PPV oligomers and single-wall carbon nanotubes toward n-type/p-type†
Juergen
Bartelmess
a,
Christian
Ehli
a,
Juan-José
Cid
b,
Miguel
García-Iglesias
b,
Purificación
Vázquez
b,
Tomás
Torres
*bc and
Dirk M.
Guldi
*a
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058, Erlangen, Germany. E-mail: guldi@chemie.uni-erlangen.de; Fax: +49 9131 8528307; Tel: +49 9131 8527341
bDepartamento de Química Orgánica, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain. E-mail: tomas.torres@uam.es; Fax: +34 91 4973966; Tel: +34 91 4975097
cIMDEA-Nanociencia. Facultad de Ciencias, Cantoblanco, 28049, Madrid, Spain
First published on 23rd December 2010
Abstract
In this work we have developed a strategy to immobilize functional groups such as ZnPc onto the surface of SWNT without, however, the needs of covalent functionalization, using substituted PPV-oligomers (1–4) with different electronic character. The complementary use of a series of microscopies and spectroscopies is central, especially to gather a comprehensive picture of the mutual interactions. Importantly, only 1, bearing electron-withdrawing CN substituents, interacts tightly with SWNT affording stable and finely dispersed SWNT suspensions. 2–4, on the other hand, fail to disperse SWNT in THF following our newly developed protocol. A likely rationale is the p-type character of PPV-oligomers 2–4, which imposes repelling forces relative to p-type SWNT, while placing CN groups yields n-type PPV-oligomer 1 and, in turn, strengthens the π–π interactions with SWNT. Photoexcitation of SWNT/1 hybrids generates a metastable radical ion pair state, namely oxidized ZnPc and reduced SWNT. Quite remarkable is the ratio of charge separation to charge recombination, namely nearly 3 orders of magnitude, which is encouraging to fabricate photovoltaic cells that are based, for example, on different nanocarbons.
Introduction
The combination of single-wall carbon nanotube (SWNT) with electron-donors or acceptors generates active materials, which are able to produce electrical energy when irradiated.1 To this end, the development of reliable and reproducible methodologies to integrate CNT into functional structures—such as donor/acceptor hybrids, able to transform sunlight into electrical or chemical energy—has emerged as an area of intense research.2 This is largely due to the unique electronic properties exhibited by these carbon allotropes and their prospects for practical applications.3 Covalent functionalization of SWNT with electron-donors either requires extensive sonication or use of strongly oxidizing agents4 and generates partial saturation of the extended π-system.5,6 As a consequence, major changes occur on the π-electronic properties and likewise their spectroscopic identity.7 An alternative strategy to control the organization between donor and acceptor units, while preserving the π-electronic structure of SWNT, is the supramolecular functionalization of SWNT by means of hydrophobic, π-stacking or van der Waals interactions with the sidewalls of SWNT.8 The non-covalent attachment of aromatic species to the SWNT surface has been widely investigated, using conjugated and non-conjugated polymers, as well as pyrene, anthracene, and porphyrins.9In terms of electron-donating materials, compounds based on macrocycles such as phthalocyanines (Pcs) represent an interesting class emanating from their ability to harvest light and subsequently occurring energy or electron transfer processes.10 Phthalocyanines with their extended electron-rich aromatic structure give rise to remarkably high extinction coefficients in the red and near infrared regions. Such features have triggered the interest in phthalocyanines in several fields including photovoltaics.11
To date, most of the examples of phthalocyanine-containing oligomers consist of cofacially stacked phthalocyanines brought together by bidentate ligands coordinated to metallophthalocyanines or silyloxy-Pc based oligomers. Less known are examples of non-conjugated12 and/or conjugated13,14 organic polymers substituted laterally by phthalocyanines. To this end, phthalocyanine moieties have been covalently linked—in the form of pending arms—to the backbone of either poly(p-phenylene vinylene) (PPV)- or polythiophene (PT)- oligomers.14 Nevertheless, subsequently performed photophysical investigations were limited to the field of molecular photovoltaics, in general,14 and the so-called “double-cable approach”,15 in particular. The use of phthalocyanine-based conjugated oligomers/polymers failed, however, to provide pertinent breakthroughs.12c A few leading examples of electron-donor/acceptor conjugates of phthalocyanines with SWNT have been described by us,16 whereas the non-covalent interactions between phthalocyanines and SWNT remain a pending issue.17
SWNT are very elusive species when it comes to the characterization of their metastable states.18 Thus, only very recently the complementary use of microscopy and spectroscopy has shed light onto mutual interactions between semiconducting SWNT and a strong electron-acceptor toward the realization of two novel milestones. The first milestone was the establishment of a versatile methodology to achieve water-soluble SWNT.19 The second milestone was the establishment of a protocol to achieve p-doped SWNT for the integration into novel optoelectronic devices.20
Furthermore, some aspects concerning the n- and p-doping that are important to, for example Fermi level engineering, are currently being studied in the field of molecular electronics—FETs,9a photovoltaics,9fetc. In this regard, the supramolecular coating of carbon nanotubes with, for instance, electron-donating polymer polyethylene imines has transformed p-type SWNT into n-type SWNTs just by changing the doping level. However, current data suggest that many other aspects of n- and p-doping still have not been assessed quantitatively by solubility surveys.
Having taken the aforementioned into account,21 we have devised, designed, and synthesized a series of phthalocyanine-based conjugated oligomers—ZnPc oligomers 1–4 (Fig. 1)—as powerful SWNT dispersants and have studied energy and charge transfer processes in the resulting SWNT/ZnPc oligomer hybrids. For the first time, particular emphasis was placed i) on exploring the relationship between the size and the strength of π-stacking interactions and ii) on demonstrating how important these aspects are for the overall stability of SWNT/ZnPc oligomer hybrids. In particular, two short PPV oligomers—n-type 1 and p-type 222—and two longer oligomers—p-type 3 and p-type 4—have been prepared and surveyed in SWNT assays.
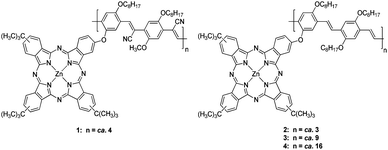 | ||
| Fig. 1 ZnPc oligomers 1–4. | ||
Results and discussion
Synthesis
Oligomer 1,22 bears cyano substituents and gives rise to strong electron-withdrawing character. The n-type character of PPV-polymers containing CN substituents is well known.23 Compound 1 was prepared by the Knoevenagel reaction, that is, reacting diformyl derivative 5 with an appropriate dicyano compound (Scheme 1). The use of high temperatures or base (i.e., Bu4NOH) triggered the Pc degradation and the partial hydrolysis of the nitrile groups, which, in turn, resulted in insoluble materials as well as low yields of low molecular weight oligomers. By the synthetic method used, only low oligomers like 1 were obtained.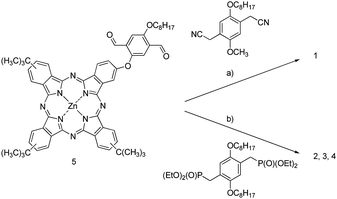 | ||
| Scheme 1 a) tBuOK (×2), Bu4NOH (×2), THF/tBuOH; 47%. b) tBuOK (×2), THF; 32% for 2, 52% for 3 and 55% for 4 —for details see Fig. 1. | ||
Oligomers 222–4 were prepared by a Wadsworth–Horner–Emmons reaction of dialdehyde 5 and 2,5-di-n-octyloxy-1,4-xylene-bis(diethylphosphonate)ester24 and potassium tert-butoxide (Scheme 1) with variable yields. In contrast to the previous case, this condensation showed a high tolerance to temperature, excess of base and time—without degradation of the oligomer and, in turn, allowing the preparation of higher molecular weight materials. Therefore, the Wadsworth–Horner–Emmons reaction emerged as an important tool to modulate the size of oligomeric materials by changing temperature of both base addition and reaction as well as reaction time. In this way, the preparation of longer oligomers 3 and 4 was accomplished by using an excess of potassium tert-butoxide (Scheme 1). For 3, the mixture of co-monomers was heated to 50 °C prior to the addition of the first batch of base. Compound 4 was synthesized in a similar way. However, unlike in the case of 3, the addition of the first batch of base was carried out at room temperature. Afterwards, both mixtures were heated for 12 h under reflux in THF followed by the addition of a second batch of base prior to extending the reaction time for another 12 h. Subsequent separation on a SEC column afforded the corresponding main fractions named as compound 3 and 4.
3 and 4, which are soluble in CHCl3, THF, and DMF, were characterized by 1H-NMR, UV-Vis, FT-IR, and GPC techniques. In general, 1H-NMR spectroscopy showed broad signals due to the presence of isomers from both Pc and PPV moieties. The signals in THF-d8 at 9.7–8.0 ppm are ascribed to the two types of hydrogen atoms surrounding the Pc core and are split into two groups. The phenyl rings of the conjugated polymer, on the other hand, are observed at 7.9–7.3 ppm. At 7.2–6.4 ppm, the vinylene linkers within the PPV backbone resonance are observed, showing the usual shift for trans-isomer vinylenes. Finally, the signals between 4.3–3.8 ppm were assigned to the methylene groups linked to the oxygen. The trans-configuration of the vinylene linkers was also confirmed by FT-IR with νδoop(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H) at 978 cm−1 in all the series.25
C–H) at 978 cm−1 in all the series.25
The GPC results in THF using polystyrene standards are summarized in Table 1. These data allow estimation of the length of the polymeric chains with regard to their average molecular weight in mass (Mw). Moreover, the average molecular weight in number, taking into account the size distributions of the chains (Mn) were determined. The quotient Mw/Mn provided insight into the regularity in size or dispersion (polydispersity, PDI). It is important to remark that in 1–4, each repetitive unit is formed by two phenylenvinylene units.
From the GPC data we established an important dependence on the polymer size (Mw, Mn) and dispersity (PDI) with respect to the reaction conditions. In particular, in 4—the oligomer with higher molecular weight—the addition of the first batch of base at room temperature, then refluxing for 12 h, followed by addition of a second batch and subsequent heating at reflux temperature for further 12 h, provided the best results.
Finally, the thermal stabilities of 2–4 were tested by thermogravimetric analyses in a nitrogen atmosphere. Thermal degradation of 2, 3, and 4 took place in two steps with different weight loss ratios. The first weight loss correlates with the degradation of the tert-butyl groups of the phthalocyanine and the PPV framework. The second weight loss is likely to be associated with the decomposition of the phthalocyanine. Within the series of 2–4, no particular trend in terms of stability and weight loss of the polymeric materials was noted. However, both the first loss of mass and the temperature of the second degradation showed clear molecular weight dependence. An immediate conclusion points to the fact that the different preparation conditions impact the number of defects and distribution thereof and, therefore, their stabilities. In any case, the overall stabilities resemble those previously described for simpler oPPV and PPV materials.26
| Mw | Mn | PDI | Size based on Mn (Repeating Units) | |
|---|---|---|---|---|
| 2 | 7211 | 5132 | 1.4 | ca. 3 |
| 3 | 21823 | 6365 | 3.429 | ca. 9 |
| 4 | 32038 | 11882 | 2.696 | ca. 16 |
Photophysics
First, the ZnPc oligomers should be considered with respect to the impact that the different PPV backbones might exert on the intrinsic ZnPc features. In the absorption spectra, 2–4 revealed the typical signatures of ZnPc, that is, maxima at around 346, 610, and 674 nm. Importantly, the features appear to be broader and red-shifted in 2 (i.e., 346, 614, and 680 nm) when compared to 3 and 4.22 In addition, the π–π* transitions of the PPV are discernible, which shift to the red from 441 nm (2) to 461 nm (4) when going from 2 to 4 as the band gap decreases with increasing length of the oligomer.In the corresponding emission spectra only the prominent features of the red emitting ZnPc evolve, without giving rise to any residual blue emission of PPV. A likely assumption is that an intramolecular energy transfer reaction governs the excited state deactivation of PPV. Complementary excitation spectra confirmed that indeed a transduction of singlet excited state energy governs the excited state chemistry of the different ZnPc oligomers.22 The quantum yields of the ZnPc emission with 0.23 for 1 and 0.18, 0.13, and 0.04 for 2–4 are, however, lower than that found in a ZnPc reference (0.3).22 We believe that different spectral overlap integrals between donor fluorescence (i.e., oligomer) and acceptor absorption (i.e., ZnPc) is responsible for this trend. Insights into the dynamics came merely from transient absorption spectroscopy, where we visualized how the oligomer excited state features transformed rapidly into those of the ZnPc singlet excited state—vide infra.
Key en-route towards the study of SWNT/ZnPc oligomers hybrids is the well-balanced preparation of semistable THF suspensions of SWNT. The latter were used to perform titration experiments—vide infra. Initially, 0.5 mg of solid SWNT were added to 3 mL of THF followed by a 10 min ultrasonication step. 3 to 4 drops of this precursor were added to 10 mL of THF followed by an additional 10 min of ultrasonication. This was repeated up to 10 times to obtain a light grayish semistable THF suspension of SWNT. Care was taken of temperature stability and careful handling of the suspension as measures to prevent aggregation/precipitation of SWNT. As a matter of fact, it is not possible to perform centrifugation with these suspensions to separate individualized SWNT from smaller and/or greater bundles. In this regard, suspensions for the following titration experiments should contain finely dispersed SWNT, for which criteria came from absorption and fluorescence spectroscopy. For example, the SWNT associated transitions as they occur between different van Hove singularities in the density of states (i.e., S22 and S11) are readily seen in the near infrared—1090, 1171, 1318, and 1455 nm.27 Attempts to map the steady-state fluorescence of SWNT with white light were without success for these diluted suspensions. Appreciable SWNT band gap fluorescence was only detected upon more intense laser excitation with, for example, a Nd/YAG laser at 532 nm. Under these experimental conditions the aforementioned absorption features (i.e., 1090, 1171, 1318, and 1455 nm) correlate well with fluorescence maxima at 1121, 1282, 1427, and 1539 nm—see Figure S1.†
Evidence for SWNT/ZnPc oligomers interactions came from absorption assays, in which the aforementioned SWNT suspensions were titrated with solutions of 1–4 in THF. Most notable is a clear bathochromic shift of the ZnPc features (i.e., Q-bands) in the visible from 675 to 693 nm for 1. Quantitatively similar are the changes for 2, 3, and 4, see Figure S2.† A significant broadening accompanies these shifts. When turning to the near infrared, the SWNT associated transitions are likewise impacted confirming that complexing SWNT to an electron-donating molecule, it was possible to modify the absorption intensity and wavelength of the SWNT transitions. In fact, distinct red-shifts and increased oscillator strengths develop throughout the titrations. Leading examples are the maxima at 876, 1214, and 1456 nm, which shift to 881, 1226, and 1463 nm. The absorption increase and red-shifting of the SWNT valence band can be attributed to electronic donation into the p-doped SWNT valence band, increasing the electronic density, while simultaneously decreasing the transition energy into the unoccupied conduction band. In the corresponding NIR fluorescence spectra—see Figure S1†—an average red shift of 14 nm evolve for the SWNT related maxima in SWNT titrated with 1–4.
In parallel fluorescence assays, emphasis was placed on the strong emission of ZnPc. The latter was found to be quenched upon addition to the SWNT suspensions without, however, giving rise to a resembling red-shift. This, in turn, leads us to hypothesize that in the free ZnPc oligomers/SWNT immobilized ZnPc oligomers equilibrium, only the former emits. Increasing the SWNT concentration perturbs the equilibrium and, as a consequence of immobilizing ZnPc oligomers onto SWNT the fluorescence intensity falls exponentially. Please note that the immobilization, at which the fluorescence quenching should be quantitative, is incomplete under these procedures. In light of the aforementioned, we estimate a fluorescence quenching of > 95% for 1 and almost complete for 2–4.
To realize the complete ZnPc oligomer immobilization onto SWNT without, however, compromising the SWNT debundling the following measures were taken. Initially, solid SWNT were added to a solution of 1. Then the resulting grayish–greenish suspensions were kept shortly—not more than 20 min—in a bath sonicator, followed by three SWNT addition/sonication cycles and centrifuged at 9.6 kG for 20 min. Importantly, ZnPc oligomers 2–4, which lack the electron-withdrawing CN groups, did not allow preparation of the stable SWNT suspensions along the aforementioned protocol at all. Two indicators were employed to monitor the progress of our procedure. Firstly, the ratio between the blue and the red absorption bands of ZnPc around 700 nm, which relate to the free and immobilized form of the ZnPc oligomers, respectively, and secondly, the resolution of the SWNT centered transitions in the 1000 to 1600 nm range—see Fig. 2.
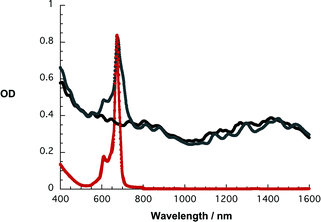 | ||
| Fig. 2 Absorption spectra of 1 in THF (red), SWNT/1 in THF (gray), and SWNT/SDBS in D2O (black). | ||
Ensuring the homogeneity of SWNT/1 is critical. Atomic force microscopy is important, since it provides important insights into this aspect. Fig. 3 confirms that throughout the scanned areas predominantly short (i.e., 5 μm) and thin bundles (i.e., 1–2 nm) are discernable.
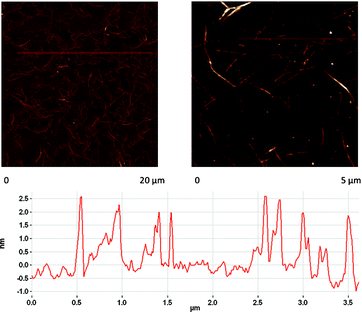 | ||
| Fig. 3 AFM images of SWNT/1 on a Si slide. Height profile derived from the image on the right hand side. | ||
Additional insight into structural and electronic aspects was derived from Raman experiments (i.e., dried on a glass slide or in solution) upon excitation at 1064 nm. In fact, the D-band in SWNT/1, which reflects structural damage of SWNT,28 is discernable at 1278 cm−1 without, however, giving rise to any notable changes when compared to SWNT/SDBS (sodium dodecylbenzenesulfate). Likewise, no changes evolved for the G-band in SWNT/1 in THF and SWNT/SDBS in D2O at 1591 cm−1 and the G′-band at 2548 cm−1.29
Debundling of SWNT was further confirmed by means of near infrared fluorescence—band gap fluorescence of individualized semiconducting SWNT—following a laser excitation at 532 nm. A leading example is shown in Fig. 4, which reveals more intense and sharper fluorescence bands, when compared to just SWNT suspensions in THF used for the titration experiments. For the latter please compare Figure S1.† Extra support for the SWNT debundling came from the fact that additional centrifugation steps fail to exert an appreciable impact on the overall fluorescence intensity. Having taken these measures into concert, we conclude the positive ZnPc oligomer immobilization and the successful SWNT debundling.30 Nevertheless, some residual ZnPc fluorescence is still seen at 680 nm.
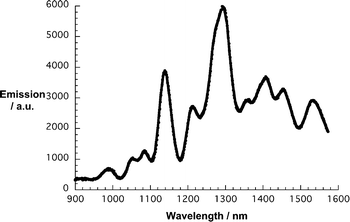 | ||
| Fig. 4 NIR fluorescence spectrum of SWNT/1 in THF upon laser excitation at 532 nm. | ||
Motivated by our success, especially in terms of debundling SWNT, we mapped the SWNT fluorescence with lamp irradiation, to probe excited state interactions. As a standard, we prepared a similar absorbing SWNT/SDBS suspension in D2O.
From Fig. 5 and 6, which compare SWNT/SDBS in D2O and SWNT/1 in THF, we derive several trends. All emission maxima at an excitation wavelength of 725 nm—assigned to (11,0), (9,4), (8,6), (8,7), and (13,3) nanotubes—are in line with the absorption spectra red-shifted. Mainly responsible are electron-donor/acceptor interactions, while sizeable contributions from solvent effects shall not be ruled out. Importantly, the fluorescence of all SWNT is on average quenched by about 80%—see Fig. 7. The strongest quenching is seen for (11,0) and (8,6) with 85% followed by (9,4) with 84%, (8,7) with 81%, and (13,3) with 78%. From the aforementioned, we conclude that no particular dependence of the quenching on the SWNT band gap emerges. We believe that the overall driving force for charge transfer, which is substantial (∼1.1 V difference between Ered(SWNT) and Eox(1)), prevents any selectivity.22,31
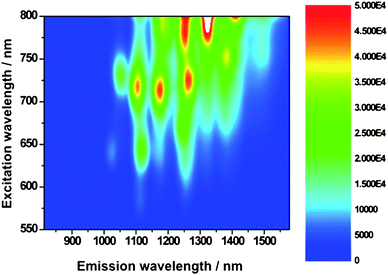 | ||
| Fig. 5 Steady-state 3D NIR fluorescence spectra of SWNT/SDBS in D2O—with increasing intensity from blue to green to yellow and to red. | ||
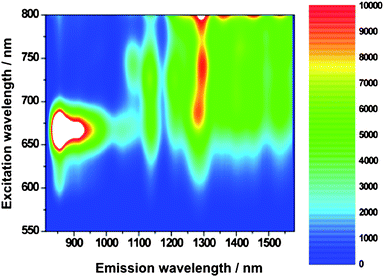 | ||
| Fig. 6 Steady-state 3D NIR fluorescence spectra of SWNT/1 in THF—with increasing intensity from blue to green to yellow and to red. | ||
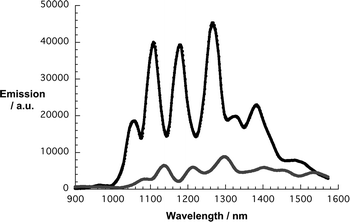 | ||
| Fig. 7 Comparison of the NIR fluorescence spectra of SWNT/SDBS in D2O (black) and SWNT/1 in THF (gray)—from Fig. 5 and 6—upon lamp excitation at 725 nm. | ||
In the final part the charge transfer postulate was corroborated by means of transient absorption measurements. In reference experiments—Fig. 8—with SWNT/SDBS suspension in D2O we note upon excitation at either 387 or 775 nm, the instantaneous bleach of the SWNT centered transitions. The latter are exact mirror images of the ground state absorption. Due to strong exciton binding, the associated excited state features are short lived and recover with complex, multi-exponential kinetics to the ground state. For the major decay components we determined lifetimes of 0.7 and 3.5 ps. Spectroscopically, the decay is a lot simpler. The recovery of the ground state is straight forward, namely not associated with any blue- or red-shift of the transient features.
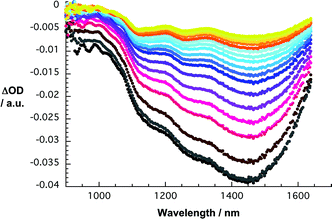 | ||
| Fig. 8 Differential absorption spectra (extended near infrared) obtained upon femtosecond flash photolysis (387 nm) of SWNT/SDBS in D2O with several time delays between 0 and 5 ps at room temperature—time evolution from black to red to blue to orange and yellow. | ||
For 1, we see the rapid formation of the singlet excited state characteristics—not shown. The latter include transient bleach in the spectral range between 600 and 700 nm. In addition, new transients are registered in the 400 to 600 nm region and around 625 nm. Fate of the ZnPc singlet excited state is an intersystem crossing (3.3 × 108 s−1) that affords the corresponding triplet manifold in about 70%.
Initially, upon photoexciting SWNT/1 at 387 nm, the ZnPc singlet excited state fingerprints—vide supra—are notable. At first glance, the visible range is dominated by transient maxima and transient minima in the 650 to 700 and 450 to 550 nm ranges, respectively. These findings are important, since they attest to the successful formation of the ZnPc singlet excited state—indirect (i.e., oligomer) or direct (i.e., ZnPc). However, a closer look in Fig. 9 at the 650 to 700 nm range reveals two features, namely a shoulder at 675 nm and a minimum at 693 nm, which resemble the free and immobilized ZnPc oligomers, respectively. Interestingly, the excited state decay of the 675 nm shoulder is a qualitative match of the slow intersystem crossing that is seen in the ZnPc oligomers. Analyzing, on the other hand, the excited state dynamics in the spectral range of the 693 nm minimum—see Fig. 10—leads to a rapid singlet excited state deactivation from which we derive a rate of ca. 1.1 × 1012 s−1. Considering approximate absorption cross sections of ZnPc and SWNT at the 387 nm excitation wavelength in a 1 to 2 ratio, a significant fraction of the light photoexcites SWNT. Therefore, we noticed—in line with reference experiments—the instantaneous bleaching of the van Hove singularities. Again, the transitions are red-shifted relative to SWNT/SDBS in D2O. In contrast to what has been seen for SWNT/SDBS in D2O the SWNT/1 decay is very fast—9.9 × 1011 s−1—and matches qualitatively that of the ZnPc singlet excited state decay immobilized onto SWNT.
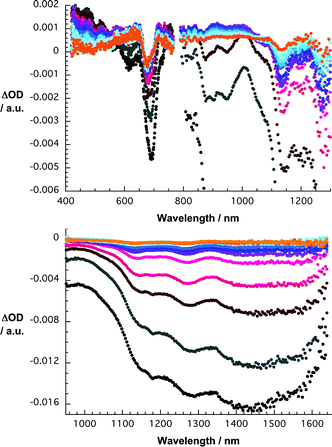 | ||
| Fig. 9 Upper part—differential absorption spectra (visible and near infrared) obtained upon femtosecond flash photolysis (387 nm) of SWNT/1 in THF with several time delays between 0 and 100 ps at room temperature—time evolution from black to red to blue and orange. Lower part —differential absorption spectra (extended near infrared) obtained upon femtosecond flash photolysis (387 nm) of SWNT/1 in THF with several time delays between 0 and 100 ps at room temperature—time evolution from black to red to blue and orange. | ||
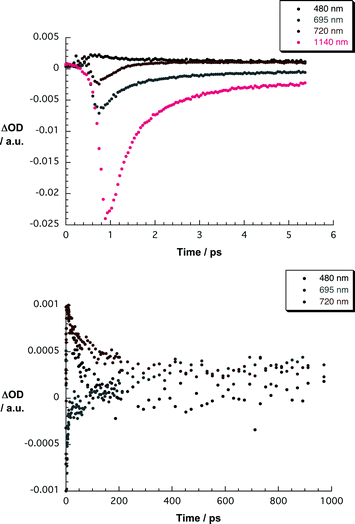 | ||
| Fig. 10 Upper part—time-absorption profiles of the spectra shown in Fig. 9 at 480, 695, 720, and 1140 nm monitoring the charge separation. Lower part—time-absorption profiles of the spectra shown in Fig. 9 at 480, 695 and 720 nm monitoring the charge recombination. | ||
At the end of the ZnPc and SWNT excited state decays, characteristic changes occur in the differential absorption for SWNT/1. In the visible, it is mainly a bleaching of the Q-bands that is appreciable. This is further accompanied by a broad absorption in the 425–550 nm range. In the near infrared, on the other hand, we see a set of maxima at 840, 920, and 1000 nm and a set of minima at 903, 947, 1141, 1180, 1270, and 1455 nm. The maxima—especially the one at 840 nm—are known to reflect the transient spectrum of the one-electron oxidized ZnPc radical cation. The same holds for the 425–550 nm transition.22,32 Notable is that a comparison of the minima with the initially generated bleach of the van Hove singularities prompts to subtle blue shifts. Implicit are new conduction band electrons—injected from photoexcited ZnPc—shifting the transitions to lower energies. Spectroscopic support for this assumption came from determining the absolute spectrum of the product and comparing it with that of the ground state. Thus, we reach the conclusion that photoexcitation of SWNT/1 hybrids is followed by a rapid charge transfer. A multi-wavelength analysis of the newly developed charge transfer state is metastable and decays with 1.9 × 109 s−1. Remarkable is—despite the overall short charge transfer lifetime—the ratio of charge separation rate to charge recombination rate of nearly 3 orders of magnitude.
Conclusions
In this work we have developed a strategy to immobilize functional groups such as ZnPc onto the surface of SWNT without, however, the needs of covalent functionalization, using substituted PPV-oligomers (1–4) with different electronic character. Among the four ZnPc oligomers (1–4) that we have probed only 1, bearing electron-withdrawing CN-substitutents, interacts tightly with SWNT affording stable and finely dispersed SWNT suspensions. ZnPc oligomers 2–4 despite having much longer lengths, failed to disperse SWNT in THF following our newly developed protocol. A likely rationale is the p-type character of PPV, in the cases of 2–4, which imposes repelling forces relative to p-type SWNT. Placing CN groups, on the other hand, onto the PPV backbone yields a n-type character PPV-oligomer and, in turn, increases π–π interactions with SWNT. The latter forces are insufficient in 2–4 to stabilize spectroscopically viable SWNT suspensions. ZnPcs are electronically “isolated” from the PPV by an ether linkage, therefore the n-type character of the CN-PPV oligomer subunit in 1 is not substantially reduced by the presence of the ZnPc units. In other words, matching the n-type character of PPV and the p-type character of SWNT is very important to immobilize with ease functional groups such as ZnPc. In the particular case of 1 other reasons for the favorable interaction with the SWNT such as a potential oligomer's unusual conformation due to CN groups and ZnPc substituents, even being less plausible, cannot be rule out. In the context of dispersion stability ZnPc seems to play a negligible role.With regards to the strong electron donor character of ZnPc, photoexcitation of SWNT/1 hybrids generates a metastable radical ion pair state, that is, the one-electron oxidized ZnPc radical cation and new SWNT conduction band electrons. Quite remarkable is the ratio of charge separation rate to charge recombination rate of nearly 3 orders of magnitude. On the other hand, in order to explore the relationship between the molecular weight of PPV-oligomers bearing electron-withdrawing groups and the strength of the stacking interactions with the SWNT, we are now preparing longer chain PPV-oligomers with the ZnPc separated from the PPV by an appropriate spacer. Currently, we are directing our attention to use this rather unique ZnPc oligomer for the fabrication of photovoltaic cells that are based on different nanocarbons.
Experimental section
Synthesis of Pc-based PPV's 3 and 4 via Wadsworth–Horner–Emmons reaction
Diformyl derivative 5 (40 mg, 0.039 mmol) and 2,5-di-n-octyloxy-1,4-xylene-bis(diethylphosphonate)ester (1.1 eq.) were dissolved in anhydrous THF (6 mL) in an argon atmosphere. Then, tBuOK (2.2 eq.) suspended in dry THF (4 mL for 3 and 4) was added dropwise under vigorous stirring to the starting monomer mixture, previously heated at 50 °C (for 3) or at room temperature (for 4). The green mixture was warmed up to the refluxing temperature for 12 h while a dark green precipitate appeared. Afterwards, additional solid tBuOK (2.2 eq.) was added in small portions and the mixture heated for further 12 h. Subsequently, when cooled down to room temperature, THF was vacuum-evaporated and the abundant green solid was triturated in an ultrasounds bath with MeOH (30 mL) acidified with a drop of AcOH. The homogenous suspension was filtered off and washed with plenty of hot MeOH, acetone and hexane until the washing solvents were colourless. The polymeric material was purified in the last step on a SEC column (Biobeads®) using freshly distilled THF as the mobile phase. The main fraction (eluting first in all the cases) was recovered and concentrated in vacuum. The polymeric material was then suspended in hot acetone, filtered and dried in vacuum, affording 3 (42 mg, 52%) and 4 (45 mg, 55%) in the form of bright dark green solids.
3: 1H- NMR (500 MHz, CDCl3), δ (ppm): 9.7–8.5 (m, 8H; PcH), 8.5–8.0 (m, 4H; PcH), 7.9–7.3 (br s, 4H; ArH), 7.2–6.4 (br s, 4H; ArCH2CH2Ar), 4.4–3.8 (m, 6H; ArOCH2), 2.2–0.6 (m, 72H; CH, CH2, CH3, C(CH3)3). UV-Vis (CHCl3), λmax (nm) 674, 611, 461, 346. FT-IR (KBr) ν (cm−1): 2959, 2932, 2864, 1611, 1492, 1393, 1331, 1287, 1261, 1220, 1194, 1155, 1099 νst(C–O–C), 1045, 978 νδ oop(CC–H trans), 925, 831, 756, 702. SEC (THF, polystyrene standards): Mw: 21823, Mn: 6365, PDI: 3.429. TGA (°C/mass lost): 368.77/−40.05%, 476.58/−6.74%.
4: 1H- NMR (500 MHz, CDCl3), δ (ppm): 9.7–8.5 (m, 8H; PcH), 8.5–8.0 (m, 4H; PcH), 7.9–7.3 (br s, 4H; ArH), 7.2–6.4 (br s, 4H; ArCH2CH2Ar), 4.4–3.8 (m, 6H; ArOCH2), 2.2–0.6 (m, 72H; CH, CH2, CH3, C(CH3)3). UV-Vis (CHCl3), λmax (nm) 674, 610, 461, 346. FT-IR (KBr) ν (cm−1): 2959, 2932, 2864, 1610, 1491, 1396, 1329, 1288, 1261, 1221, 1194, 1153, 1099 νst(C–O–C), 1045, 978 νδ oop(CC–H trans), 924, 831, 756, 702, 534, 451. SEC (THF, polystyrene standards): Mw: 32038, Mn: 11882, PDI: 2,696. TGA (°C/mass lost): 387.74/−32.64%, 466.70/−11.67%.
Photophysical measurements
THF was of spectrophotometric grade (99.5%) and was purchased from Sigma–Aldrich. SWNTs (HiPCO Batch Nr.: R0510C) were purchased from CNI. The experiments were performed at room temperature and ambient conditions. Steady-state absorption spectra were measured by a Cary5000 (Varian) two beam spectrometer. Emission spectra were recorded by using a FluoroMax-P (HORIBA Jobin Yvon). NIR emission spectra were measured by a Fluorolog spectrometer (HORIBA Jobin Yvon). Here, the optical detection was performed by a Symphony InGaAs array in combination with an iHR320 imaging spectrometer. The samples were excited by a 450 W Xenon lamp or the SHG (532 nm) of a Nd/YAG laser. Femtosecond transient absorption studies were performed using 387 and 775 nm laser pulses with a 150 fs pulse width (1 kHz, 200 nJ), from an amplified Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sapphire laser system (SHG and fundamental, CPA 2001, Clark-MXR Inc.) additional excitation experiments was carried by using 660 and 700 nm pulses created by a NOPA (Clark-MXR Inc.). For the detection we used a Helios TAPPS from Ultrafast Inc. AFM images were recorded on a Solver Pro scanning probe microscope (NT-MDT Co). The carbon nanotube material was spread on oxidized silicon wafers by spin casting. Raman spectra were recorded using a Bruker FT Raman RFS 100 system with a liquid N2 cooled Ge detector upon excitation by a 1064 nm Nd–YAG laser.
:sapphire laser system (SHG and fundamental, CPA 2001, Clark-MXR Inc.) additional excitation experiments was carried by using 660 and 700 nm pulses created by a NOPA (Clark-MXR Inc.). For the detection we used a Helios TAPPS from Ultrafast Inc. AFM images were recorded on a Solver Pro scanning probe microscope (NT-MDT Co). The carbon nanotube material was spread on oxidized silicon wafers by spin casting. Raman spectra were recorded using a Bruker FT Raman RFS 100 system with a liquid N2 cooled Ge detector upon excitation by a 1064 nm Nd–YAG laser.
Acknowledgements
We are grateful to Eva M. Maya for GPC and TGA measurements. Financial support from the MICINN and MEC (Spain) (CTQ2008-00418/BQU, PLE2009-0070, and Consolider-Ingenio Nanociencia Molecular CSD2007-00010), Comunidad de Madrid (MADRISOLAR-2, S2009/PPQ/1533) and EU (MRTN-CT-2006-035533 Solar-N-type and Project ROBUST DSC FP7-Energy-2007-1-RTD, N° 212792) is gratefully acknowledged. We are grateful to the Zentralinstitut für Neue Materialien und Prozesstechnik in Fürth for the possibility to perform AFM measurements and the Deutsche Forschungsgemeinschaft (SFB 583 and Clusters of Excellence Engineering of Advanced Materials).Notes and references
- (a) D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871 CrossRef CAS; (b) D. M. Guldi, G. M. A. Rahman, V. Sgobba, N. A. Kotov, D. Bonifazi and M. Prato, J. Am. Chem. Soc., 2006, 128, 2315 CrossRef CAS; (c) V. Sgobba, G. M. A. Rahman, D. M. Guldi, N. Jux, S. Campidelli and M. Prato, Adv. Mater., 2006, 18, 2264 CrossRef.
- (a) C. Ehli, G. M. A. Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222 CrossRef CAS; (b) R. Chitta, A. S. D. Sandanayaka, A. L. Schumacher, L. D'Souza, Y. Araki, O. Ito and F. D'Souza, J. Phys. Chem. C, 2007, 111, 6947 CrossRef CAS; (c) A. Kongkanand, R. M. Domínguez and P. V. Kamat, Nano Lett., 2007, 7, 676 CrossRef CAS.
- (a) S. Iijima, Nature, 1991, 354, 56 CrossRef CAS; (b) H. Dai, Acc. Chem. Res., 2002, 35, 1035 CrossRef CAS.
- (a) K. C. Hwang, J. Chem. Soc., Chem. Commun., 1995, 2, 173 Search PubMed; (b) D. B. Mawhinney, V. Naumenko, A. Kuznetzova and J. T. Yates Jr, J. Am. Chem. Soc., 2000, 122, 2383 CrossRef CAS; (c) S. Banerjee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17 CrossRef CAS.
- (a) Y. Saito and S. Uemura, Carbon, 2000, 38, 169 CrossRef CAS; (b) S. Bellucci, Phys. Status Solidi C, 2005, 2, 34 Search PubMed.
- (a) W. Huang, Y. Lin, S. Taylor, J. Gaillard, A. M. Rao and Y.-P. Sun, Nano Lett., 2002, 2, 231 CrossRef CAS; (b) A. Lucas, C. Zakri, M. Maugey, M. Pasquali, P. van der Schoot and P. Puolin, J. Phys. Chem. C, 2009, 113, 20599 CrossRef CAS.
- (a) A. Kuznetsova, I. Popova, J. T. Yates Jr, M. J. Bronikowski, M. C. B. Huffman, J. Liu, R. E. Smalley, H. H. Hwu and J. G. Chen, J. Am. Chem. Soc., 2001, 123, 10699 CrossRef CAS; (b) J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712 CrossRef CAS; (c) S. Osswald, E. Flahaut and Y. Gogotsi, Chem. Mater., 2006, 18, 1525 CrossRef CAS.
- (a) D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS; (b) H. Murakami and N. Nakashima, J. Nanosci. Nanotechnol., 2006, 6, 16 CAS; (c) A. Star, Y. Liu, K. Grant, L. Ridvan, J. F. Stoddart, D. W. Steuerman, M. R. Diehl, A. Boukai and J. R. Heath, Macromolecules, 2003, 36, 553 CrossRef CAS.
- (a) M. Shim, A. Javey, N. W. S. Kam and H. Dai, J. Am. Chem. Soc., 2001, 123, 11512 CrossRef CAS; (b) J. Chen, H. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034 CrossRef CAS; (c) N. Nakashima, Y. Tomonari and H. Murakami, Chem. Lett., 2002, 31, 638 CrossRef; (d) J. Zhang, J.-K. Lee, Y. Wu and R. W. Murray, Nano Lett., 2003, 3, 403 CrossRef CAS; (e) H. Murakami, T. Nomura and N. Nakashima, Chem. Phys. Lett., 2003, 378, 481 CrossRef CAS; (f) Z. Li, V. Saini, E. Dervishi, V. P. Kunets, J. Zhang, Y. Xu, A. R. Biris, G. J. Salamo and A. S. Biris, Appl. Phys. Lett., 2010, 96, 033110 CrossRef.
- (a) T. Nojiri, M. M. Alam, H. Konami, A. Watanabe and O. Ito, J. Phys. Chem. A, 1997, 101, 7943 CrossRef CAS; (b) D. M. Guldi, I. Zilberman, A. Gouloumis, P. Vázquez and T. Torres, J. Phys. Chem. B, 2004, 108, 18485 CrossRef CAS; (c) A. R. M. Soares, M. V. Martínez-Díaz, A. Bruckner, A. M. V. M. Pereira, J. P. C. Tomé, C. M. A. Alonso, M. A. F. Faustino, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva, J. A. S. Cavaleiro, T. Torres and D. M. Guldi, Org. Lett., 2007, 9, 1557 CrossRef CAS; (d) G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000 RSC.
- (a) J. Xue, S. Uchida, B. P. Rand and S. R. Forrest, Appl. Phys. Lett., 2004, 84, 3013 CrossRef CAS; (b) K. Suemori, T. Miyata, M. Yokoyama and M. Hiramoto, Appl. Phys. Lett., 2005, 86, 063509 CrossRef; (c) J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martínez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS; (d) B. E. Hardin, E. T. Hoke, P. B. Armstrong, J.-H. Yum, P. Comte, T. Torres, J. M. J. Frechet, M. K. Nazeeruddin, M. Grätzel and M. D. McGehee, Nat. Photonics, 2009, 3, 406 CrossRef CAS; (e) Ballesteros, G. de la Torre, T. Torres, G. L. Hug, G. M. A. Rahman and D. M. Guldi, Tetrahedron, 2006, 62, 2097 CrossRef CAS; (f) M. V. Martínez-Díaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090 RSC.
- (a) M. Kimura, K. Wada, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Macromolecules, 2001, 34, 4706 CrossRef CAS; (b) A. S. Drager, R. A. P. Zangmeister, N. R. Amstrong and D. F. O'Brien, J. Am. Chem. Soc., 2001, 123, 3595 CrossRef CAS; (c) A. de la Escosura, M. V. Martínez-Díaz, T. Torres, R. H. Grubbs, D. M. Guldi, H. Neugebauer, C. Winder, M. Drees and N. S. Sariciftci, Chem.–Asian J., 2006, 1, 148 CrossRef CAS.
- (a) F. Zamora, M. C. González and J. J. del Val, J. Macromol. Sci., Part B: Phys., 1998, 37, 601 Search PubMed; (b) P. Zhao, Y. Song, S. Dong, L. Niu and F. Zhang, Dalton Trans., 2009, 6327 RSC; (c) M. Hanack and P. Stihler, Eur. J. Org. Chem., 2000, 303 CrossRef CAS; (d) M. Kimura, H. Narikawa, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Chem. Mater., 2002, 14, 2711 CrossRef CAS; (e) I. Akai, H. Nakao, K. Kanemoto, T. Karasawa, H. Hashimoto and M. Kimura, J. Lumin., 2005, 112, 449 CrossRef CAS.
- M. V. Martínez, S. Esperanza, A. de la Escosura, M. Catellani, S. Yunus, S. Luzzati and T. Torres, Tetrahedron Lett., 2003, 44, 8475 CrossRef CAS.
- (a) A. Cravino and N. S. Sariciftci, J. Mater. Chem., 2002, 12, 1931 RSC; (b) A. Cravino and N. S. Sariciftci, Nat. Mater., 2003, 2, 360 CrossRef CAS; (c) H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125 CrossRef CAS; (d) J. Roncali, Chem. Soc. Rev., 2005, 34, 483 RSC; (e) A. J. Mozer and N. S. Sariciftci, C. R. Chim., 2006, 9, 568 CrossRef CAS; (f) Z. Tan, J. Hou, Y. He, E. Zhou, C. Yang and Y. Li, Macromolecules, 2007, 40, 1868 CrossRef CAS; (g) A. Cravino, Polym. Int., 2007, 56, 943 CrossRef CAS.
- (a) B. Ballesteros, S. Campidelli, G. de la Torre, C. Ehli, D. M. Guldi, M. Prato and T. Torres, Chem. Commun., 2007, 2950 RSC; (b) B. Ballesteros, G. de la Torre, C. Ehli, G. M. A. Rahman, F. Agulló-Rueda, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2007, 129, 5061 CrossRef CAS; (c) S. Campidelli, B. Ballesteros, A. Filoramo, D. D. Díaz, G. de la Torre, T. Torres, G. M. A. Rahman, C. Ehli, D. Kiessling, F. Werner, V. Sgobba, D. M. Guldi, C. Cioffi, M. Prato and J. Bourgoin, J. Am. Chem. Soc., 2008, 130, 11503 CrossRef CAS.
- (a) R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838 CrossRef CAS; (b) C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2007, 8, 75; (c) R. A. Hatton, N. P. Blanchard, V. Stolojan, A. J. Miller and S. R. P. Silva, Langmuir, 2007, 23, 6424 CrossRef CAS; (d) S. Kyatskaya, J. R. G. Mascarós, L. Bogani, F. Hennrich, M. Kappes, W. Wernsdorfer and M. Rubén, J. Am. Chem. Soc., 2009, 131, 15143 CrossRef CAS; (e) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS.
- (a) C. Ehli, D. M. Guldi, M. Á Herranz, N. Martín, S. Campidelli and M. Prato, J. Mater. Chem., 2008, 18, 1498 RSC; (b) M.Á. Herranz, C. Ehli, S. Campidelli, M. Gutiérrez, G. L. Hug, K. Ohkubo, S. Fukuzumi, M. Prato, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2008, 130, 66 CrossRef; (c) C. Ehli, C. Oelsner, D. M. Guldi, A. Mateo-Alonso, C. Schmidt, C. Backes, F. Hauke and A. Hirsch, Nat. Chem., 2009, 1, 243 CrossRef CAS.
- (a) M. J. O'Connell, P. Boul, L. M. Ericson, E. Haroz, C. Kuper, J. Tour, K. D. Ausman and R. E. Smalley, Chem. Phys. Lett., 2001, 342, 265 CrossRef CAS; (b) M. F. Islam, E. Rojas, D. M. Bergey, A. T. Johnson and A. G. Yodh, Nano Lett., 2003, 3, 269 CrossRef CAS; (c) Y. Sabba and E. L. Thomas, Macromolecules, 2004, 37, 4815 CrossRef CAS; (d) Y. Liu, L. Gao, S. Zheng, Y. Wang, J. Sun, H. Kajiura, Y. Li and K. Noda, Nanotechnology, 2007, 18, 365702 CrossRef.
- (a) H. S. Woo, R. Czerw, S. Webster, D. L. Carroll, J. Ballato, A. E. Strevens, D. O'Brien and W. J. Blau, Appl. Phys. Lett., 2000, 77, 1391; (b) A. Javey, M. Shim and H. Dai, Appl. Phys. Lett., 2002, 80, 1064 CrossRef CAS; (c) D. M. Guldi, G. M. A. Rahman, N. Jux, N. Tagmatarchis and M. Prato, Angew. Chem., Int. Ed., 2004, 43, 5526 CrossRef CAS; (d) G. M. A. Rahman, A. Troeger, V. Sgobba, D. Balbinot, M. N. Tchoul, W. T. Ford, A. Mateo-Alonso and M. Prato, Chem.–Eur. J., 2008, 14, 8837 CrossRef CAS; (e) V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165 RSC.
- (a) V. H. Crespi, M. L. Cohen and A. Rubio, Phys. Rev. Lett., 1997, 79, 2093 CrossRef CAS; (b) P. Petit, C. Mathis, C. Journet and P. Bernier, Chem. Phys. Lett., 1999, 305, 370 CrossRef CAS; (c) J. Lee, H. Kim, S.-J. Khang, G. Kim, Y.-W. Son, J. Ihm, H. Kato, Z. W. Wang, T. Okazaki, H. Shinohara and Y. Kuk, Nature, 2002, 415, 1005 CrossRef CAS; (d) E. D. Minot, Y. Yaish, V. Sazonova, J.-Y. Park, M. Brink and P. L. McEuen, Phys. Rev. Lett., 2003, 90, 156401 CrossRef CAS; (e) Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273 CrossRef CAS.
- J.-J. Cid, C. Ehli, C. Atienza-Castellanos, A. Gouloumis, E.-M. Maya, P. Vázquez, T. Torres and D. M. Guldi, Dalton Trans., 2009, 3955 RSC.
- (a) N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. C. Friend and A. B. Holmes, Nature, 1993, 365, 628 CrossRef; (b) J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS; (c) G. Yu and A. J. Heeger, J. Appl. Phys., 1995, 78, 4510 CrossRef CAS; (d) S. C. Moratti, R. Cervini, A. B. Holmes, D. R. Baigent, R. H. Friend, N. C. Greenham, J. Grüner and P. J. Hamer, Synth. Met., 1995, 71, 2117 CrossRef CAS; (e) M. M. de Souza, G. Rumbles, D. L. Russell, I. D. W. Samuel, S. C. Moratti, A. B. Holmes and P. L. Burn, Synth. Met., 2001, 119, 635 CrossRef CAS; (f) Y. Liu, G. Yu, Q. Li and D. Zhu, Synth. Met., 2001, 122, 401 CrossRef CAS; (g) D. A. M. Egbe, T. Kietzke, B. Carbonnier, D. Mühlbacher, H.-H. Hörhold, D. Neher and T. Pakula, Macromolecules, 2004, 37, 8863 CrossRef CAS; (h) S. W. Chasteen, S. A. Carter and G. Rumbles, J. Chem. Phys., 2006, 124, 214704 CrossRef CAS; (i) V. Boucard, Macromolecules, 2001, 34, 4308 CrossRef CAS; (j) M. Hanack, B. Behnisch, H. Häckl, P. Martinez-Ruiz and K.-H. Schweikart, Thin Solid Films, 2002, 417, 26 CrossRef CAS; (k) J.-P. Morin, N. Drolet, Y. Tao and M. Leclerc, Chem. Mater., 2004, 16, 4619 CrossRef CAS; (l) P. Teranekar, M. Abdulbaki, R. Krishnamoorti, S. Phanichphant, P. Waenkaew, D. Patton, T. Fulghum and R. Advicula, Macromolecules, 2006, 39, 3848 CrossRef.
- A. Drury, S. Meier, M. Rüther and W. J. Blau, J. Mater. Chem., 2003, 13, 485 RSC.
- (a) K. F. Voss, C. M. Foster, L. Smilowitz, D. Mihailovic, S. Askari, G. Srdanov, Z. Ni, S. Shi, A. J. Heeger and F. Wudl, Phys. Rev. B: Condens. Matter, 1991, 43, 5109 CrossRef CAS; (b) S. Pfeiffer and H.-H. Hörhold, Macromol. Chem. Phys., 1999, 200, 1870 CrossRef CAS.
- F. Wudl and S. Hoger, US Pat., 5679757, 1997.
- (a) S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361 CrossRef CAS; (b) M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS; (c) R. B. Weisman and S. M. Bachilo, Nano Lett., 2003, 3, 1235 CrossRef CAS; (d) A. Hartschuh, H. N. Pedrosa, L. Novotny and T. D. Krauss, Science, 2003, 301, 1354 CrossRef CAS; (e) D. Yoon, S.-J. Kang, J.-B. Choi, Y.-J. Kim and S. Baik, J. Nanosci. Nanotechnol., 2007, 7, 3727 CrossRef CAS.
- (a) P. C. Eklund, J. M. Holden and R. A. Jishi, Carbon, 1995, 33, 959 CrossRef CAS; (b) M. S. Dresselhaus, A. Jorio, A. G. Souza Filho, G. Dresselhaus and R. Saito, Physica B, 2002, 15 Search PubMed; (c) G. Chen, G. U. Sumanasekera, B. K. Pradhan, R. Gupka, P. C. Eklund, M. J. Bronikowski and R. E. Smalley, J. Nanosci. Nanotechnol., 2002, 2, 621 CrossRef CAS; (d) M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47 CrossRef; (e) S. Lefrant, I. Baltog and M. Baibarac, Synth. Met., 2009, 159, 2173 CrossRef CAS.
- (a) I. O. Maciel, N. Anderson, M. A. Pimento, A. Hartschuh, H. Qian, M. Terrones, H. Terrones, J. Campos-Delgado, A. M. Rao, L. Novotny and A. Jorio, Nat. Mater., 2008, 7, 878 CrossRef CAS; (b) M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751 CrossRef CAS.
- (a) C. D. Spataru, S. Ismail-Beigi, L. X. Benedict and S. G. Louie, Phys. Rev. Lett., 2004, 92, 077402 CrossRef; (b) M. J. O'Connell, E. E. Eibergen and S. K. Doorn, Nat. Mater., 2005, 4, 412 CrossRef CAS; (c) T. Hertel, A. Hagen, V. Talalaev, K. Arnold, F. Hennrich, M. Kappes, S. Rosenthal, J. McBride, H. Ulbricht and E. Flahaut, Nano Lett., 2005, 5, 11 CrossRef CAS.
- (a) K. Iakoubovskii, N. Minami, S. Kazaoui, T. Ueno, Y. Miyati, K. Yanagi, H. Kautura, S. Ohshima and T. Saito, J. Phys. Chem. B, 2006, 110, 17420 CrossRef CAS; (b) J. Lefebvre and P. Finnie, Nano Lett., 2008, 8, 1890 CrossRef CAS; (c) J. Wang and Y. Li, J. Am. Chem. Soc., 2009, 131, 5364 CrossRef CAS; (d) Y. Tanaka, Y. Hirana, Y. Niidome, K. Kato, S. Saito and N. Nakashima, Angew. Chem., Int. Ed., 2009, 48, 7655 CrossRef CAS.
- (a) T. Nyokong, Z. Gasyna and M. J. Stillman, Inorg. Chem., 1987, 26, 548 CrossRef; (b) M. E. El-Khouly, O. Ito, P. M. Smith and F. D'Souza, J. Photochem. Photobiol., C, 2004, 5, 79 CrossRef CAS; (c) A. Kahnt, M. Quintiliani, P. Vázquez, D. M. Guldi and T. Torres, ChemSusChem, 2008, 1, 97 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NIR fluorescence and absorption spectra of SWNT titrated with 1 in THF. See DOI: 10.1039/c0sc00364f |
| This journal is © The Royal Society of Chemistry 2011 |