N-Aryl-substituted 3-(β-D-glucopyranosyloxy)-2-methyl-4(1H)-pyridinones as agents for Alzheimer's therapy†
Lauren E.
Scott
a,
Maria
Telpoukhovskaia
a,
Cristina
Rodríguez-Rodríguez
a,
Michael
Merkel
a,
Meryn L.
Bowen
a,
Brent D. G.
Page
a,
David E.
Green
a,
Tim
Storr
a,
Fancy
Thomas
b,
David D.
Allen
c,
Paul R.
Lockman
b,
Brian O.
Patrick
a,
Michael J.
Adam
d and
Chris
Orvig
*a
aMedicinal Inorganic Chemistry Group, Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC, Canada V6T 1Z1. E-mail: orvig@chem.ubc.ca; Tel: +1 604 822 4449
bSchool of Pharmacy, Texas Tech University Health Sciences Center, 1300 Coulter, Suite 112, Amarillo, TX 79106, USA
cNortheastern Ohio Universities College of Pharmacy, Rootstown, OH 44272, USA
dTRIUMF, 4004 Wesbrook Mall, Vancouver, BC, Canada V6T 2A3
First published on 23rd December 2010
Abstract
Molecules designed to sequester, redistribute and/or remove metal ions are attractive therapeutic agents in neurodegenerative diseases such as Alzheimer's disease. The multifactorial nature of the condition and the generally poor target specificity associated with metal ion-binding therapy has led to the development of multifunctional 3-hydroxy-4-(1H)-pyridinone pro-ligands. The excellent qualities of the basic 3-hydroxy-4-pyridinone framework as a low toxicity metal chelator and an antioxidant, as well as its antibacterial and analgesic properties among other functions, inspired us to functionalize it with a framework derived from thioflavin-T, the well-known traditional dye used as a marker to detect amyloid deposits in tissue sections. Thus 2-methyl-3-hydroxy-1-(4-dimethylaminophenyl)-4(1H)-pyridinone (HL1), 2-methyl-3-hydroxy-1-(4-methylaminophenyl)-4(1H)-pyridinone (HL2), 1-(4-aminophenyl)-3-hydroxy-2-methyl-4(1H)-pyridinone (HL3), 1-(6-benzothiazolyl)-3-hydroxy-2-methyl-4(1H)-pyridinone (HL4), 1-(2-benzothiazolyl)-3-hydroxy-2-methyl-4(1H)-pyridinone (HL5) and 2-methyl-3-hydroxy-1-[4-(4-bromophenyl)-2-thiazolyl]-4(1H)-pyridinone (HL6) were obtained. Glycosylation, as well as incorporation of structures mimicking those of known amyloid imaging agents, may target drug action to the site of interest, the metal-overloaded amyloid plaques in the Alzheimer's brain. The pro-ligands were assessed for their antioxidant activity, cytotoxicity and ability to interfere with metal ion-induced amyloid peptide aggregation to screen promising lead compounds. Finally, in a brain uptake study with a radiolabeled glucoconjugate pyridinone, 3-(β-D-glucopyranosyloxy)-1-[4-(4-[125I]iodophenyl)-2-thiazolyl]-2-methyl-4(1H)-pyridinone ([125I]-GL7) was shown to cross the blood–brain barrier using an in situ rat brain perfusion technique.
Introduction
Alzheimer's disease (AD) is the most common cause of age-related senile dementia. It affects more than 24 million people worldwide, with this number expected to reach over 81 million by 2040.1 Two biochemical features of AD contributing to neurodegeneration are intracellular oxidative stress and elevated levels of trace metal ions, especially Fe3+, Cu2+ and Zn2+ in certain areas of brain tissue.2,3 These are associated with the pathological markers of AD, extracellular β-amyloid plaques and intracellular neurofibrillary tangles,4–7 which are typically used for post-mortem AD diagnosis and are linked in a coherent manner by the amyloid hypothesis.8 In an effort to reduce the harmful effects of excess metal ions and oxidative stress, therapies involving metal-ion chelation and/or removal are a promising therapeutic approach for AD.9,10 Systemic chelators like desferrioxamine (DFO), clioquinol (CQ, both Fig. 1) or clioquinol's analog PBT2 (having recently completed a phase IIa clinical trial)11 have been tested in murine AD models and AD patients.9,12,13 CQ has been shown to dissolve Alzheimer-like amyloid deposits in transgenic mice and in some cases to slow the cognitive decline associated with AD.12,14 PBT2 has been shown to lower levels of Aβ1–42 in the CSF of AD patients without affecting serum metal ion levels (Cu2+, Zn2+) and appears to be well tolerated.11 Metal chelation agents provide one such function to adjust localized metal imbalances in the brain; however, enhancing the targeting and efficacy of these chelators' design is a significant part of the strategy in the development of the next generation of multifunctional agents.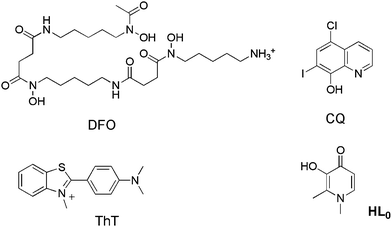 | ||
| Fig. 1 Deferrioxamine (DFO), clioquinol (CQ), thioflavin-T (ThT) and deferiprone (1,2-dimethyl-3-hydroxy-4(1H)-pyridinone), herein called HL0. | ||
In addition to metal chelating ability, these agents also contain features designed to improve their uptake across the blood–brain barrier (BBB) as well as multiple functionalities to better treat the many factors occurring simultaneously in AD. A more in-depth discussion of the use of metal chelators in AD therapy has already been published.15
On the basis of the above premises, the development of metal chelators with potential use in AD is the main objective of this work. The strategy consists of creating small multifunctional molecules that contain structural moieties for metal chelation and Aβ recognition/interaction. For the first purpose, the basic 3-hydroxy-4-pyridinone framework of deferiprone (HL0, Fig. 1) is an excellent starting point for rational drug development because of its high affinity for Cu(II), as well as Zn(II), low affinity for common biological electrolyte ions such as sodium, potassium, calcium and magnesium, its antioxidant properties and low toxicity.16 In addition to these properties, the structure is amenable to functionalization by N-substituent variation, which can alter the physical properties of the drug without significant alteration of metal ion binding.17 Some noteworthy molecules containing 3-hydroxy-4-pyridinones possess antibacterial,18 antimalarial,19 antineoplastic,20 anti-inflammatory,21 analgesic21,22 and oral metal chelating23 activities. In some countries in Europe, HL0 is in clinical use as an oral chelator to mitigate iron overload in thalassemia patients.24 For the latter purpose, we incorporate a framework derived from thioflavin-T (ThT, Fig. 1), the well-known traditional dye used as a marker to detect amyloid deposits in tissue sections because of its strong affinity for amyloid fibrils, similar approaches have been described by other groups.25–28 Notwithstanding the fact that ThT has been used for several decades, it is known that it is not specific for amyloids of Aβ, as it binds to many types of amyloid. Thus, the molecular understanding of this interaction is still an object of intensive study, which is critical in order to design new dyes to bind amyloid fibrils more specifically and for the development of diagnostic and therapeutic agents.29,30 For this reason, three different motifs derived from the ThT have been chosen to be incorporated into the molecular framework depicted in Fig. 2.
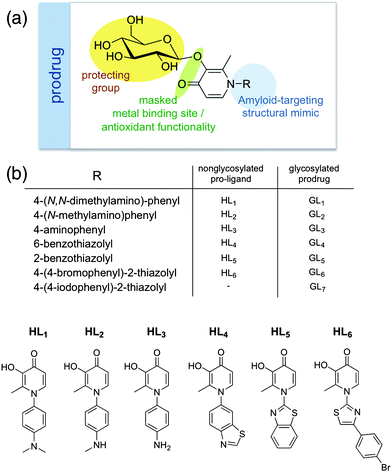 | ||
| Fig. 2 (a) Rational scaffold designed for multifunctional masked pyridinone prodrug for AD therapy. (b) Generalized structures and names of all pyridinone pro-ligands and glycosylated prodrugs discussed in this work. | ||
We were also interested in functionalizing these molecules by glycosylation in order to increase prodrug solubility31 and according to our previous work32 it would also improve the pharmacokinetics by preventing metabolism and excretion. Furthermore, it could mitigate the undesired chelation of systemic metal ions. The glycoconjugate has been used by various groups to impart elevated brain uptake by exploitation of the glucose transport system in the blood–brain barrier, with glycosylated compounds exhibiting improved uptake into the brain.33–35
As shown previously, once the glycosylated prodrug (prodrug or GLn) is enzymatically cleaved by the β-glucosidase enzymes (present in high levels in the brain) the desired active form of the drug (pro-ligand or HLn) is released.36 This active form is able to bind metals (becoming a ligand or M(Ln)x) and exhibit antioxidant activity.
Herein, we report the design, synthesis, and characterization of novel multifunctional pyridinone pro-ligands that include (1) glycosylation as part of the prodrug approach to improve BBB penetration of the compound and access to the brain and masking, once the free pro-ligand is exposed by glycosidic bond hydrolysis, the active drug includes activities such as (2) metal ion complexation, (3) phenolic H-donation for antioxidant activity and (4) Aβ imaging agent structural mimicry designed to localize the compound to the site of Aβ deposition. Additionally, since these molecules closely match Lipinski's rules,37 they fulfil the drug-like criteria for further biological in vivo assays. All the functions outlined in the design have been verified and the corresponding data is presented in the this work.
Results and discussion
Pyridinone pro-ligands
The pro-ligands are prepared in three different steps: first the 3-hydroxy functional group is protected as either a methyl- or benzyl-ether generating 3-methoxy- or 3-benzoloxy-2-methyl-4-pyranone. Although in theory the conversion of pyranone to pyridinone can be achieved without any protection at the 3-hydroxyl group,38 this reaction tends to be extremely low-yielding. This protected pyranone is reacted with the appropriate primary amine, typically under extreme conditions such as high temperature (up to 120 °C) and over an extended period of time (up to 96 h). The protected pyridinone is isolated by column chromatography. Finally, the protecting group is removed under acidic conditions to yield the unprotected compound. Precipitation from an acidic or a basic solution containing the compound by neutralization yields the purified free pro-ligand target molecule. Before the chelating and biological properties were evaluated, the structures of five pro-ligands (HL1, HL2, HL3, HL4 and HL5) were established by X-ray diffraction in order to analyze their structural properties (see Fig. S1 and Tables S1–S2 for details†). The main features of the molecular structures are very similar to those of the parent 3-hydroxy-4-pyridinone compounds determined by our group and others.39–42Glycosylated prodrugs
Although some work has been done with pyridinone formation from O-glycosylated pyranone precursors,43 there are few precedents for O-glycosylation of 3-hydroxy-4-pyridinones. To perform the functionalization significant variations from previous works were needed.32 Briefly, the deprotonation of the 3-hydroxy-pyridinone in dry methanolic KOH and the addition to 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide yielded consistent results for O-glycosylation of all pyridinones; in addition, the basic conditions allowed removal of the carbohydrate acetyl protection groups. The general synthetic scheme was modified for the preparation of 1-(4-aminophenyl)-3-(β-D-glucopyranosyloxy)-2-methyl-4(1H)-pyridinone (GL3). In this case, a precursor, 3-hydroxy-2-methyl-1-(4-nitrophenyl)-4(1H)-pyridinone (10c) was glycosylated instead; the presence of a nitrosyl functional group allowed glycosylation to occur at the 3-hydroxyl position of the pyridinone without addition of a glycosyl group on the primary amino group of HL3. This modified route is outlined in Scheme 1. Thin layer chromatography (TLC) easily monitored the progression of glycosylation reactions. Additionally, for the synthesis of the iodinated compound GL7, the four sugar hydroxyl groups in GL6 were reacetylated with acetic anhydride and the bromo substituent was converted into a tributylstannyl group to give 12. Finally, the tributylstannyl group was exchanged for an iodo group (9f) and the acetyl protecting groups removed under basic conditions to give GL7 (Scheme 1).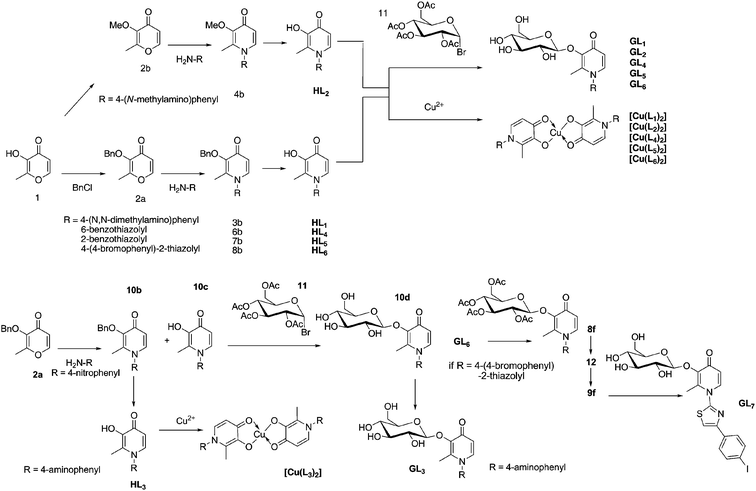 | ||
| Scheme 1 General scheme for the synthesis of pyridinones, pyridinone glycosides and Cu(II) complexes | ||
Bis(pyridonato)copper(II) complexes
The neutral Cu2+ complexes were synthesized by deprotonation of pyridinone pro-ligand with triethylamine and reaction with copper(II) perchlorate. In all reactions, the solid complex precipitated out from solution as a light green powder that afforded good quality single crystals suitable for X-ray diffraction. All the complexes were characterized by electrospray ionization-MS, elemental analysis, infrared spectroscopy and for those that crystallized, by X-ray diffraction. According to the crystal structure data obtained, the Cu2+ ion resides on a center of inversion resulting in a symmetric molecule around it; all complexes display nearly square planar coordination spheres about the metal ion and trans-ligand geometries (Fig. S2, ESI†). Since all six copper complexes have similar geometry around the copper centre, the X-band EPR spectrum of a powder sample for one representative complex [Cu(L1)2] was recorded. Although X-ray diffraction data for a number of pyridinone complexes of Fe(III), Al(III), Ga(III) and In(III) have been reported,42,44–46 only a few examples of Cu(II) complexes are available.47 The EPR study was used to confirm the speciation of copper in the bis(pyridinato) complexes. The α-hydroxyketone moiety of the pyridinone ligand is known to coordinate Cu2+ in solution even in the presence of other possible donor atoms such as those in amino or carboxylate groups.48 Previous studies on bis(maltolato)copper(II) complexes and bis(L-mimosinato) complexes (both containing the α-hydroxyketo-chelating moiety) were characterized by EPR and shown to contain Cu2+ centers in axial coordination environments, as expected for square-planar bis-bidentate ligand complexes.48 The EPR spectrum obtained (Fig. S3, ESI†) contains a g⊥-value typical of a Cu2+ center with a (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, O−) donor set at 2.063 (compared to that of maltol, L-mimosine Cu2+ complexes at 2.304).48 The expected hyperfine (HF) splitting peaks are observed, arising from coupling of the lone electron with the Cu2+ center (d9, nuclear spin = 3/2). It should be noted that although four peaks are expected, only three are clearly discernable on the spectrum, as one HF peak overlaps the main spectral line. This EPR experiment does confirm the solution speciation of the copper ion in these bis(pyridinato) complexes as Cu2+.
O, O−) donor set at 2.063 (compared to that of maltol, L-mimosine Cu2+ complexes at 2.304).48 The expected hyperfine (HF) splitting peaks are observed, arising from coupling of the lone electron with the Cu2+ center (d9, nuclear spin = 3/2). It should be noted that although four peaks are expected, only three are clearly discernable on the spectrum, as one HF peak overlaps the main spectral line. This EPR experiment does confirm the solution speciation of the copper ion in these bis(pyridinato) complexes as Cu2+.
The overall stability constants (log β) for the copper complexes formed by deferiprone (herein called HL0) are 10.62 (log β1) and 19.61 (log β2) at 25 °C and 0.10 M KCl ionic strength.42,47,49 From the speciation diagram the stoichiometry of the existing species at pH 6.6 are [Cu(L0)]+ and [Cu(L0)]2, the latter being predominant. Moreover, the concentration of free Cu(II) ions above pH 4 is negligible. Since variations of the N-substituent of pyridinones is known not to significantly modulate metal binding activity,17 it is expected that HL1–HL6 will have similar speciation in the physiological regime.
Drug-like properties of the pro-ligands and prodrugs
Drug-like filters, such as the Lipinski's rules,37 are very helpful for assessing the likelihood that a given molecule could be bio-available. These simple rules37 state that bio-availability is likely to occur if at least three of the following rules are obeyed: molecular weight is less than 500, calculated logarithm of the octanol–water partition coefficient (clogP) is less than 5, the number of hydrogen-bond donors (HBD) is less than 5; the number of hydrogen-bond acceptors (HBA) is less than 10.37 It is significant that the complete series of glycosylated prodrug and non-glycosylated pro-ligands closely match the Lipinski rule of five37 (see ESI†).Moreover, these druglikeness filters reflect indirectly that a good drug candidate should show both a reasonable aqueous solubility and its ability to diffuse passively through the different biological barriers (lipophilicity and H-bond capabilities cutoffs, respectively). However, excessively polar compounds would have difficulty at the stage of passing through the various biological barriers such as the BBB. Many small molecules, such as nicotine, cross the BBB via passive diffusion.50 This transport of a molecule across the cell membrane is determined by its molecular size, charge, lipophilicity, and hydrogen bonding capability. In order to evaluate whether or not the molecules of this work could pass the BBB by passive diffusion, we calculated the log BB parameter, a common measure of the degree of BBB penetration, using the equation log BB = −0.0148 TPSA + 0.152 clogP + 0.139.51,52 This equation has shown good predictive ability and comprises of two simple variables that can be rapidly computed for any structure: the calculated logarithm of the octanol–water partition coefficient (clogP) and the topological polar surface area (TPSA). The structures and log BB values are shown in Fig. S4 in the ESI†. The main criteria for determining if a molecule could pass the BBB is the log BB value greater than −0.3.25 As a result, based on the calculated log BB parameter as a computational measure of passive diffusion alone, one would expect that only HL6 passes the BBB, additionally if the Br is replaced by I, the log BB (−0.24) is also favorable.
Carrier-mediated transport may be also active, in which ATP is consumed upon transport, and is normally unidirectional, or may be facilitative, which is driven by a concentration gradient, and can function in either direction. Glucose, which is the major energy source for the brain, is facilitatively transported via the glucose transporter (GLUT-1) through various membranes. This transporter is known to be expressed in high concentrations at the BBB and in erythrocytes and the fact that the compound tested was a glycosyl-linked prodrug, it is plausible that the glycosylated compounds enter the brain through carrier-mediated transport. Recently, studies evaluated the ability of HL0 (Fig. 1) and its glucosylated analogue to cross the BBB by means of in situ brain perfusion technique53 finding that HL0 was able to penetrate BBB in guinea pigs, whereas its glucosylated analogue was unable to do so. These two molecules were also added to our calculations for comparison of results (Fig. S4, ESI†). From the calculated log BB parameter we may conclude that since HL0 is a small drug-like compound and has a log BB value close to the cutoff, it may be able to passively diffuse through membranes or use other transport mechanisms rather than carrier-mediated transport. On the contrary, the glycosylated GL0 is not able to pass through the BBB via passive transport according to the calculated log BB (Fig. S4, ESI†). This is supported by the experimental data.53 In the same work,53 the conclusion that the glycosylation cannot be relied upon as a general means of enhancing the brain uptake of this molecular class should take into account a series of related compounds rather than be based only on two pyridinones (HL0 and GL0).
Radioiodination and rat brain uptake assay
One representative gycosylated pyridinone, GL6, was selected for its availability and was tested for radiolabeling and rat brain uptake assay‡. 125I was introduced to the N-aryl-substituent on the pyridinone rather than to the carbohydrate part of the target molecule, since the glucoside glycosidase enzymes in vivo may cleave that linkage and the biodistribution of the sugar may differ from that of the pyridinone. Brain uptake of a radiolabeled glucoconjugate pyridinone was assessed using the same in situ rat brain perfusion technique54 used in previous pyridinone studies reported by our group.32 Briefly, radiolabeled [125I]-GL7 in saline was perfused for 60 s into the common carotid artery, the animal was terminated, and the brain radioactivity assessed to determine tracer BBB penetration. In order to compare the pyridinone compound ([125I]-GL7) against a few references of known brain permeation, a plot was constructed of observed BBB permeation vs. calculated octanol–water partition coefficient at pH 7.4 (ClogD) (Fig. S5, ESI†). Sucrose crosses the BBB very slowly, and for this reason it is commonly used as a negative control. On the contrary, caffeine and nicotine pass through the BBB easily and they have been used as positive controls. Nicotine is a very small molecule (MW 162 g mol−1), and crosses the BBB via passive diffusion. Caffeine (MW 194 g mol−1), while also small, is a structural analog of the purine base adenosine, and known to cross the BBB via both passive diffusion and carrier-mediated transport.50,55 Our test compound, [125I]-GL7, falls somewhere in the middle of these two extremes, with a calculated permeability of 0.9 ± 0.4 μL s−1g−1, similar to thiourea, although not as high as caffeine, theophylline or ethanol (values determined previously).56 Data indicate that since the brain uptake of [125I]-GL7 is lower than that expected from passive diffusion through membranes, other transport mechanisms such as carrier-mediated transport (GLUT-1) may take place.Antioxidant activity of pro-ligands
Oxidative stress is implicated in Alzheimer's disease; redox cycling of metal ions (e.g. Cu2+) associated with the Aβ peptide can produce reactive oxygen species (ROS) such as the extremely reactive hydroxyl radical, HO˙, that can abstract hydrogen atoms thereby causing oxidative damage to lipids, proteins, DNA, etc. With the aim of assessing the antioxidant activity of the pro-ligands HL1–HL5, the Trolox equivalent antioxidant capacity (TEAC) assay was used to quantify it.57 This assay examines the quenching of 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS˙+) radical cation decolorization in presence of antioxidant compounds by means of UV-vis spectroscopy.The ability of HL1–HL5 to quench the ABTS˙+ radical cation was compared to Trolox, a more water-soluble analog of α-toco-pherol, and the results are shown in Fig. 3. All of the pyridinone pro-ligands tested, with the exception of HL5, possibly due to its low solubility, showed comparable antioxidant capacity to α-tocopherol and exceed that of butylated hydroxytoluene (BHT), both used as positive controls. It is clear from this study that HL1–HL5 pro-ligands have the capability to act as antioxidant compounds contributing to the multifunctional nature of these potential AD therapeutics.
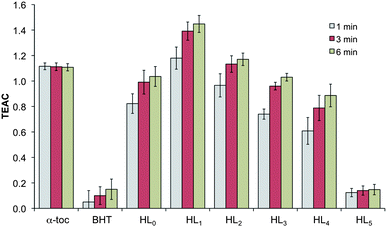 | ||
| Fig. 3 TEAC values at 1, 3, and 6 min for known antioxidants α-Toc (α-tocopherol) and BHT (butylated hydroxytoluene), and tested compounds. Means are the average of three independent trials, and error bars show standard deviations. | ||
Cytotoxicity studies
Human hepatocellular liver carcinoma cells (line HepG2) were used in this study to determine the toxicity of two representative pro-ligands (HL3 and HL4) in comparison to the cytotoxic agent cisplatin (cis-diammine-dichloroplatinum(II)).The selected cells are adequate to study in this work as these are cultured in a microcosm for studying drug effect on the liver, the organ responsible for drug metabolism and detoxification.58 The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay59 which is an in vitro colorimetric method of determining cell viability was used here to assess cell toxicity. The IC50 values of the tested compounds were 92 ± 18 μM and 19 ± 5 μM for compounds HL3 and HL4, respectively. These values are above or on the order of observed IC50 value for cisplatin (15 ± 3 μM), the reference compound. Based on these and previous results of pyridinone cytotoxicities by the MTT assay,60 it seems that more hydrophilic pyridinone N-substituents, such as that in HL3, tend to imbue pyridinones with higher IC50 values (lower toxicity). These two representative pro-ligands show acceptably low cytotoxicity, such that the application of these and similar pyridinones as metal chelating drugs is feasible when low concentrations are used. This initial screen suggests that a toxicity study of the full series of these compounds, and their glycosylated analogues, is warranted in a more relevant cell model.
Aβ aggregation inhibition studies
Two representative pyridinones, HL3 and HL4, were tested for their ability to resolubilize metal-aggregated Aβ1–40 species via a turbidimetry assay. For the assay, zinc(II) and copper(II) ions, which are known to cause Aβ aggregation in solution,28,61 were added to synthetic human Aβ1–40 dissolved in a buffered aqueous solution that was assessed at physiological pH (7.4) for Zn2+ and at lower pH (6.6) for Cu2+. The formation of the aggregates leads to increased light scattering, which was verified by increased solution turbidity as measured at 405 nm prior to addition of the test compound. Within this context, depletion of the cations from solution by complexation with a reference chelator such as diethylenetriaminepentaacetic acid (DTPA) and/or HL0 (Fig. 1), prevents most of the ion-promoted aggregation effect. The two compounds studied in this work (HL3 and HL4) are able to reduce Aβ1–40 peptide aggregation in the presence of Cu(II) and Zn(II) (Fig. 4). They clearly decrease fibril buildup in the presence of Cu2+ with similar resolubilization efficacy as DTPA. On the contrary, for Zn2+ trials, DTPA and HL0 were significantly more effective for Aβ disaggregation than any of the pyridinone pro-ligands assayed. It is apparent that the structure of the pro-ligand does have some bearing on resolubilization efficacy in the case of Zn2+-aggregated Aβ1-40.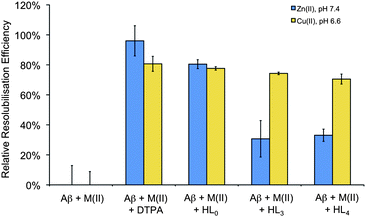 | ||
| Fig. 4 Relative resolubilization efficiency towards Cu(II) or Zn(II) mediated Aβ1–40 aggregates of HL3 and HL4 as compared to HL0 and DTPA (normalized to 100). | ||
The observation of a differential effect on Cu(II)- and Zn(II)-promoted aggregation might suggest that these compounds are not acting as non-specific chelators but rather recognize a subpopulation of Cu(II) cations involved in the self-assembly of the peptide. Indeed, it has been reported that different mechanisms lie behind Zn(II)- and Cu(II)-induced aggregation.2 In the case of copper, its redox chemistry is involved in the origin of the ROS species that are implicated in the pathogenesis of AD.2 Disruption of these aberrant metal–peptide interactions via chelation therapy could thus be considered as a promising therapeutic strategy for combating this disease.2,15
Concerning the design of the multifunctional prodrug, the trends observed herein may indicate that while the pyridinone structure may be advantageous in this application, the structural nature of N-substituent aryl group does not seem to affect ligand propensity for interaction with metal-Aβ binding site(s) or reduction of metal ion-induced Aβ aggregation. Developments in this line are currently underway in order to assess influences on the affinity towards Aβ aggregates of the exocyclic nitrogen substituent, as well as the benzotiazol ring, functionalizing them with different groups in terms of bulk and hydrophobicity. Further experiments will be required to understand the dominant processes involved in the interaction of pyridinones with metal ion-aggregated Aβ peptide, but the current results highlight the potential benefit of molecular structural variation to affect pyridinone biological function.
Conclusions
Incorporation of structural moieties for Aβ recognition/interaction into a well-known 3-hydroxy-4-pyridinone framework of deferiprone chelator has been shown to be successful in the design of multifunctional agents for Alzheimer's disease. Following this strategy in this work six novel pro-ligands (HL1–HL6) have been prepared and characterized by mass spectrometry, 1H and 13C NMR spectroscopies, elemental analysis and, with the exception of HL6, X-ray crystallography. To demonstrate the feasibility of the prodrug approach, the glycosylated analogs of all of them have been synthesized and characterized as well. The coordination features of the synthesized ML2 [M = Cu(II); L = L1, L3, L6] complexes were characterized and for [Cu(L1)2] also analyzed by EPR confirming consistent ion coordination, as expected, via the α-hydroxyketone functional group of the pyridinone ring. The pro-ligands displayed significant antioxidant activity similar to or better than α-tocopherol and acceptably low toxicity. According to the turbidity assays, two representative pro-ligands, HL3 and HL4, can be efficient chelators for sequestering the Cu(II) and Zn(II) metal ions that are present in soluble Aβ peptide forms, especially in the case of Cu(II). Finally, a brain uptake study with a radiolabeled glucoconjugate pyridinone, [125I]-GL7, indicated adequate cerebral uptake of this prodrug, establishing the initial validity of brain-permeating pyridinones as AD therapeutics.Overall, these preliminary findings show that hydroxypyridinone pro-ligands with embued antioxidant, metal chelation, and Aβ dissociation properties may be developed into treatments for AD patients. All functions outlined when tested in isolation worked and since two analogues cross the BBB, the design is ready to move into biochemical and in vivo studies.
Acknowledgements
The authors gratefully acknowledge the Canadian Institutes of Health Research (CIHR) for a Proof of Principle grant. LES recognizes support from the Alzheimer Society of Canada and the Institute of Aging (CIHR) for a doctoral training award, the Province of British Columbia for a Pacific Century Scholarship, and the University of British Columbia for a University Graduate Fellowship. MM thanks the Alexander von Humboldt Foundation for a Feodor Lynen-Fellowship. CRR wishes to express her gratitude for the financial support received from the Agencia de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) from Generalitat de Catalunya for her grant Beatriu de Pinós (2009 BP-A 00056). MT recognizes support from the Alzheimer Society of Canada for a doctoral award. We also thank Dr. Dana Devine and her lab for allowing us the use of their plate reader.References
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Lancet, 2005, 366, 2112–2117.
- A. I. Bush, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS.
- R. D. Moir, C. S. Atwood, D. M. Romano, M. H. Laurans, X. Huang, A. I. Bush, J. D. Smith and R. E. Tanzi, Biochemistry, 1999, 38, 4595–4603 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609 CrossRef CAS.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS.
- D. R. Crapper McLachlan, A. J. Dalton, T. P. Kruck, M. Y. Bell, W. L. Smith, W. Kalow and D. F. Andrews, Lancet, 1991, 337, 1304–1308 CrossRef.
- R. A. Cherny, J. T. Legg, C. A. McLean, D. P. Fairlie, X. Huang, C. S. Atwood, K. Beyreuther, R. E. Tanzi, C. L. Masters and A. I. Bush, J. Biol. Chem., 1999, 274, 23223–23228 CrossRef CAS.
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2008, 7, 779–786 CrossRef CAS.
- C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi and C. L. Masters, Arch. Neurol., 2003, 60, 1685–1691 CrossRef.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. S. Kim, X. D. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS.
- E. J. Gralla, R. B. Stebbins, G. L. Coleman and C. S. Delahunt, Toxicol. Appl. Pharmacol., 1969, 15, 604–613 CrossRef CAS.
- P. S. Dobbin, R. C. Hider, A. D. Hall, P. D. Taylor, P. Sarpong, J. B. Porter, G. Xiao and D. van der Helm, J. Med. Chem., 1993, 36, 2448–2458 CrossRef CAS.
- M.-H. Feng, L. van der Does and A. Bantjes, J. Med. Chem., 1993, 36, 2822–2827 CrossRef CAS.
- C. Hershko, E. N. Theanacho, D. T. Spira, H. H. Peter, P. Dobbin and R. C. Hider, Blood, 1991, 77, 637–643 CAS.
- F. Timeus, P. Valle, N. Crescenzio, L. Ruggieri, P. Rosso, G. Luca Pagliardi, L. Cordero di Montezemolo, V. Gabutti and U. Ramenghi, Am. J. Hematol., 1994, 47, 183–188 CrossRef CAS.
- G. Ozturk, D. D. Erol, M. D. Aytemir and T. Uzbay, Eur. J. Med. Chem., 2002, 37, 829–834 CrossRef CAS.
- M. D. Aytemir, T. Uzbay and D. D. Erol, Drug Res., 1999, 49, 250–254 Search PubMed.
- G. J. Kontoghiorghes, M. A. Aldouri, L. Sheppard and A. V. Hoffbrand, Lancet, 1987, 1, 1294–1295 CrossRef CAS.
- R. Galanello, Ther. Clin. Risk Manage., 2007, 3, 795–805 Search PubMed.
- C. Rodríguez-Rodríguez, N. Sánchez de Groot, A. Rimola, A. Alvarez-Larena, V. Lloveras, J. Vidal-Gancedo, S. Ventura, J. Vendrell, M. Sodupe and P. González-Duarte, J. Am. Chem. Soc., 2009, 131, 1436–1451 CrossRef CAS.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS.
- W.-h. Wu, P. Lei, Q. Liu, J. Hu, A. P. Gunn, M.-s. Chen, Y.-f. Rui, X.-y. Su, Z.-p. Xie, Y.-F. Zhao, A. I. Bush and Y.-m. Li, J. Biol. Chem., 2008, 283, 31657–31664 CrossRef CAS.
- A. Dedeoglu, K. Cormier, S. Payton, K. A. Tseitlin, J. N. Kremsky, L. Lai, X. H. Li, R. D. Moir, R. E. Tanzi, A. I. Bush, N. W. Kowall, J. T. Rogers and X. D. Huang, Exp. Gerontol., 2004, 39, 1641–1649 CrossRef CAS.
- C. Rodríguez-Rodríguez, A. Rimola, L. Rodríguez-Santiago, P. Ugliengo, A. Álvarez-Larena, H. Gutiérrez-de-Teran, M. Sodupe and P. González-Duarte, Chem. Commun., 2010, 46, 1156–1158 RSC.
- R. L. Yona, S. Mazeres, P. Faller and E. Gras, ChemMedChem, 2008, 3, 63–66 CrossRef.
- B. Gyurcsik and L. Nagy, Coord. Chem. Rev., 2000, 203, 81–149 CrossRef CAS.
- H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, B. O. Patrick, K. H. Thompson and C. Orvig, Angew. Chem., Int. Ed., 2007, 46, 1716–1718 CrossRef CAS.
- C. Fernandez, O. Nieto, J. A. Fontenla, E. Rivas, M. L. de Ceballos and A. Fernandez-Mayoralas, Org. Biomol. Chem., 2003, 1, 767–771 RSC.
- G. Battaglia, M. La Russa, V. Bruno, L. Arenare, R. Ippolito, A. Copani, F. Bonina and F. Nicoletti, Brain Res., 2000, 860, 149–156 CrossRef CAS.
- E. J. Bilsky, R. D. Egleton, S. A. Mitchell, M. M. Palian, P. Davis, J. D. Huber, H. Jones, H. I. Yamamura, J. Janders, T. P. Davis, F. Porreca, V. J. Hruby and R. Polt, J. Med. Chem., 2000, 43, 2586–2590 CrossRef CAS.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- K. Imafuku, K. Takahashi and H. Matsumura, Bull. Chem. Soc. Jpn., 1979, 52, 111–113 CrossRef CAS.
- W. O. Nelson, T. B. Karpishin, S. J. Rettig and C. Orvig, Can. J. Chem., 1988, 66, 123–131 CAS.
- Z. Zhang, S. J. Rettig and C. Orvig, Can. J. Chem., 1992, 70, 763–770 CAS.
- D. E. Green, C. L. Ferreira, R. V. Stick, B. O. Patrick, M. J. Adam and C. Orvig, Bioconjugate Chem., 2005, 16, 1597–1609 CrossRef CAS.
- E. T. Clarke, A. E. Martell and J. Reibenspies, Inorg. Chim. Acta, 1992, 196, 177–183 CrossRef CAS.
- M. Yasue, N. Kawamura and J. Sakakibara, Yakugaku Zasshi, 1970, 90, 1222–1225 CAS.
- J. Charalambous, A. Dodd, M. McPartlin, S. O. C. Matondo, N. D. Pathirana and H. R. Powell, Polyhedron, 1988, 7, 2235–2237 CrossRef CAS.
- Z. Zhang, S. J. Rettig and C. Orvig, Inorg. Chem., 1991, 30, 509–515 CrossRef CAS.
- B. L. Ellis, A. K. Duhme, R. C. Hider, M. B. Hossain, S. Rizvi and D. van der Helm, J. Med. Chem., 1996, 39, 3659–3670 CrossRef CAS.
- A. El-Jammal, P. L. Howell, M. A. Turner, N. Li and D. M. Templeton, J. Med. Chem., 1994, 37, 461–466 CrossRef CAS.
- E. Chruscinska, E. Garribba, G. Micera and A. Panzanelli, J. Inorg. Biochem., 1999, 75, 225–232 CrossRef CAS.
- V. M. Nurchi, G. Crisponi, T. Pivetta, M. Donatoni and M. Remelli, J. Inorg. Biochem., 2008, 102, 684–692 CrossRef CAS.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS.
- D. E. Clark, J. Pharm. Sci., 1999, 88, 815–821 CrossRef CAS.
- J. T. Goodwin and D. E. Clark, J. Pharmacol. Exp. Ther., 2005, 315, 477–483 CrossRef CAS.
- S. Roy, J. E. Preston, R. C. Hider and Y. M. Ma, J. Med. Chem., 2010, 53, 5886–5889 CrossRef CAS.
- Q. R. Smith and D. D. Allen, Meth. Mol. Med., 2003, 89, 209–218 Search PubMed.
- A. L. McCall, W. R. Millington and R. J. Wurtman, Life Sci., 1982, 31, 2709–2715 CrossRef CAS.
- Q. R. Smith, Meth. Mol. Med., 2003, 89, 193–208 Search PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS.
- N. B. Javitt, FASEB J., 1990, 4, 161–168 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- L. E. Scott, B. D. G. Page, B. O. Patrick and C. Orvig, Dalton Trans., 2008, 6364–6367 RSC.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, M. E. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817–11282 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Electronic supplementary information (ESI) available: Full experimental details for the synthesis, spectroscopic and crystallographic characterization of HL1–HL5 and Cu(II) complexes, druglikeness and log BB parameter calculations. Experimental details for antioxidant studies, Aβ1 − 40 turbidity, cytotoxicity (MTT) and in vivo brain uptake assays are also described. CCDC reference numbers 798377–798384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00544d |
| ‡ All studies were approved by the Animal Care and Use Committee at Texas Tech University Health Sciences Center, and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals. |
| This journal is © The Royal Society of Chemistry 2011 |