Probing surface site heterogeneity through 1D and INADEQUATE31P solid state NMR spectroscopy of silica supported PMe3-Au(I) adducts†
David
Gajan
abc,
Daniel
Levine
a,
Eva
Zocher
ac,
Christophe
Copéret
*ac,
Anne
Lesage
b and
Lyndon
Emsley
*b
aUniversité de Lyon, Institut de Chimie de Lyon, C2P2 UMR 5265 (CNRS – CPE – Université Lyon 1), ESCPE Lyon, 43, Bd. du 11 Novembre, F-69616, Villeurbanne, France
bUniversité de Lyon, Centre de RMN à Très Hauts Champs, CNRS/ENS-Lyon/UCB-Lyon 1. 5 rue de la Doua, 69100, Villeurbanne, France. E-mail: lyndon-emsley@ens-lyon.fr
cETH Zürich, Department of Chemistry, Wolfgang-Pauli-Str. 10, CH-8093, Zürich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch
First published on 21st February 2011
Abstract
Grafting of (Me3Si)2N-AuPMe3 on SiO2-(700) leads to the formation of [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)Au-PMe3] located in different surface environments due to the presence or absence of adjacent siloxane bridges, as shown both by 31P solid state NMR and by its conversion into a mixture of [(SiO)Au-PMe3] and [(SiO)Au(PMe3)2] upon exposure to PMe3.
SiO)Au-PMe3] located in different surface environments due to the presence or absence of adjacent siloxane bridges, as shown both by 31P solid state NMR and by its conversion into a mixture of [(SiO)Au-PMe3] and [(SiO)Au(PMe3)2] upon exposure to PMe3.
Detailed characterization of surface species is critical when trying to develop heterogeneous catalystsvia a structure-reactivity relationship approach.1–6 This strategy has been successfully applied to the development of highly efficient “single-site” heterogeneous catalysts, e.g. in alkene polymerisation2 and metathesis.7,8 Well-defined surface species with controlled site density can also be used to control the growth and formation of nanoparticles on supports.9,10 A recent example is the formation of small Au nanoparticles (1.8 nm), with a narrow size distribution on passivated silica, which display very good activity for the aerobic epoxidation of Z-stilbene and for the preferential oxidation of CO (PROX) in the presence of H2.10 In that work, we prepared and characterized at a molecular level well-defined Au(I) surface species, [(
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) SiO)Au-L] {L = NH3-x(SiMe3)x}, by reaction of [AuN(SiMe3)2]4 on silica partially dehydroxylated at 700 °C (SiO2-(700)). Surprisingly, we observed the formation of both mono- and bis-PMe3 adducts upon contact of this species with an excess of PMe3 (Scheme 1a).
SiO)Au-L] {L = NH3-x(SiMe3)x}, by reaction of [AuN(SiMe3)2]4 on silica partially dehydroxylated at 700 °C (SiO2-(700)). Surprisingly, we observed the formation of both mono- and bis-PMe3 adducts upon contact of this species with an excess of PMe3 (Scheme 1a).
![Grafting and further reaction with PMe3 of a) [AuN(SiMe3)2]410 and b) (Me3Si)2NAuPMe3 on SiO2-(700).](/image/article/2011/SC/c0sc00579g/c0sc00579g-s1.gif) | ||
| Scheme 1 Grafting and further reaction with PMe3 of a) [AuN(SiMe3)2]410 and b) (Me3Si)2NAuPMe3 on SiO2-(700). | ||
The objective of this work is to understand the structure of Au(I) surface species and their formation by investigating the reaction of (Me3Si)2N-AuPMe3 and SiO2-(700), and by studying the reactivity of the resulting surface species with PMe3 (Scheme 1b). Here, we show the direct spectroscopic evidence of surface heterogeneity for well-defined silica supported Au(I) surface species through 1D and INADEQUATE 31P NMR spectroscopy.
First, the reaction of SiO2-(700) with varying amounts of (Me3Si)2N-AuPMe3 (X = 0.5, 0.75, 1.0 and 2.0 equiv/surface silanol) in pentane yielded white solids of general formula {Au(PMe3)/SiO2}X containing increasing amounts of Au with a constant Au/P ratio of 1, but reaching a ratio of 1.5 Au per surface silanol for X = 2.0 (i.e., an excess of Au per surface silanol) despite extensive washing (Table 1). Such an observation is rare, as in most cases no more than one metal is grafted per surface silanol,1,11 suggesting the formation of polynuclear species at higher loading (X = 2.0).
| Number of equiv. of Au per surface silanola | Elemental Analysis | ||
|---|---|---|---|
| % Au | Au/SiOH | % P (P/Au) | |
| a 0.26 mmol g−1 of SiO2-(700). | |||
| X = 0.5 | 2.4 ± 0.1 | 0.5 ± 0.1 | 0.4 (1) |
| X = 0.75 | 2.8 ± 0.1 | 0.6 ± 0.1 | 0.5 (1) |
| X = 1.0 | 4.4 ± 0.1 | 0.9 ± 0.1 | 0.7 (1) |
| X = 2.0 | 7.7 ± 0.1 | 1.5 ± 0.1 | 1.2 (1) |
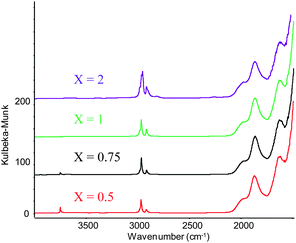
Analysis of these solids by Diffused Reflectance Infrared Fourier Transform spectroscopy (DRIFT) shows an increasing consumption of surface silanols (3700 cm−1) with increasing gold loading. Note that almost complete grafting (> 95%) is achieved with a loading of 0.75 equivalents, suggesting the passivation of some of the silanols as OSiMe3 surface species (Fig. 1).
In 1H solid-state magic angle spinning (MAS) NMR spectra, shown in Fig. 2a, two peaks are observed at 0.0 and 1.6 ppm, corresponding to Si(CH3)3 and P(CH3)3, respectively, the remaining surface OH being buried under the signal of PMe3. Note the increasing amount of PMe3vs.Si(Me)3 with increasing loading. At low loadings (X ≤ 1) only two peaks are observed in 13C CPMAS (Cross Polarization Magic Angle Spinning) NMR spectra, at 0 and 16.4 ppm, which can be assigned to OSi(CH3)3 and P(CH3)3, respectively (Fig. 2b). While a single resonance is observed for coordinated PMe3 in the 13C NMR spectra, two very close resonances are observed in 31P CPMAS NMR spectra at −17.3 and −12.1ppm in a roughly 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 2c), which can both be assigned to [(SiO)Au-PMe3] surface species.
:1 ratio (Fig. 2c), which can both be assigned to [(SiO)Au-PMe3] surface species.
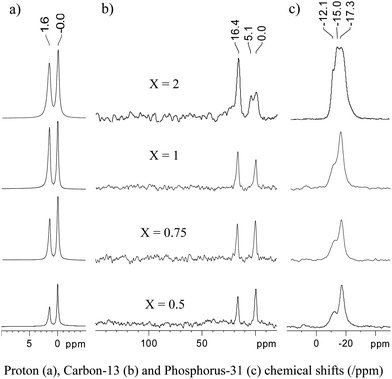 | ||
| Fig. 2 Solid-state NMR spectra of the product resulting from the grafting of {Au(PMe3)/SiO2}X onto partially dehydroxylated silica. All spectra were recorded on a 500 MHz Bruker Avance III spectrometer, equipped with a double resonance 4 mm MAS probe. The spinning frequency was 10 kHz. a) 1H single pulse spectra (8 scans with a recycle delay of 16 s). b) 13C CPMAS spectra. A total of 6800 scans were accumulated with a recycle delay of 8 s. Cross-polarization was achieved using a linear ramp of the rf field amplitude (100% to 90%) on the 1H channel, with a 2 ms CP contact time and a 1H rf field strength of ω1H/2π = 72 kHz. For the 13C channel, the rf field strength was adjusted for optimum transfer efficiency. SPINAL-64 heteronuclear decoupling was applied during acquisition (ω1H/2π ∼ 84 kHz). c) 31P CPMAS spectra. A total of 1340 scans were accumulated with a recycle delay of 5 s. The contact time for CP was 5 ms. Other experimental conditions are the same as for b). | ||
This inequivalence is probably associated with species in slightly different environments resulting from surface heterogeneity (vide infra). At higher loading (X = 2.0), new peaks appear in both the 13C and 31P NMR spectra. Considering the presence of 1.5 equiv of Au per surface silanol according to elemental analysis, and the amount of this species that remains despite extensive washing, we interpret this as the formation of a dinuclear Au species. However, it has so far not been possible to obtain further information on the nature of these dinuclear species.
To further characterise these systems and to compare them with our previous data resulting from the adsorption of PMe3 on gold(I) surface species,10 we investigated the reaction of PMe3 on {Au(PMe3)/SiO2}X and examined the resulting material by 31P NMR (Fig. 3a).It is noteworthy that in all cases the signal at −17.6 ppm remains while the signals previously observed at lower field (−12.3 ppm as well as the peak at −15 for X =2) disappear.
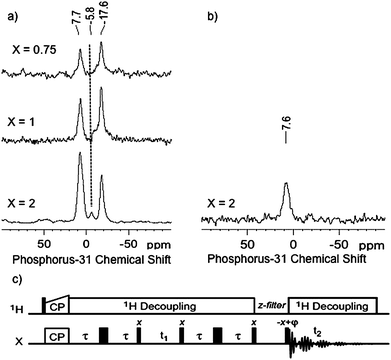 | ||
| Fig. 3 a) 31P CPMAS NMR spectra of the {Au(PMe3)/SiO2}X materials after reaction with PMe3. All spectra were recorded on a 500 MHz Bruker Avance III spectrometer, equipped with a double resonance 2.5 mm MAS probe. The sample spinning frequency was 12.5 kHz. Each spectrum was acquired by accumulating 1024 scans with a recycle delay between scans of 5 s. Cross-polarization was achieved using a linear ramp of rf field amplitude on the 1H channel (100% to 90%), with a 5 ms CP contact time and a 1H rf field strength of ω1H/2π = 72 kHz. For the 31P channel, the rf field strength was chosen for optimum transfer efficiency. SPINAL-64 heteronuclear decoupling was applied during acquisition (ω1H/2π ∼ 84 kHz). b) One-dimensional 31P refocused INADEQUATE spectrum of {Au(PMe3)/SiO2}2 after reaction with PMe3. A total of 10240 scans were accumulated with a recycle delay of 2 s. The delay τ was set to 3 ms for the two echo periods and a z-filter delay of 3 ms was used before acquisition. c) the z-filtered refocused (zfr) INADEQUATE pulse sequence used.12a | ||
Moreover, their disappearance is associated with the appearance of a new peak at 7.7 ppm, which has previously been attributed to a bis(phosphine) adduct, [Au(PMe3)2+], for [(SiO)Au-L] treated with an excess of PMe3.10 An extra signal of weak intensity for X = 2 is also observed at −5.8 ppm, which can be tentatively attributed to HOPMe3+, resulting from the presence of trace amounts of adventitious O=PMe3. Note also a slight increase of line broadening from ca. 1000 to 1200 Hz for [Au(PMe3)2+] upon increasing Au loading, which is probably indicating the presence of different local environments. A refocused INADEQUATE (Incredible Natural Abundance Double QUAntum Transfer Experiment) experiment12 is a method of choice to identify pairs of bonded nuclei, including when the two nuclei have the same isotropic chemical shift.13 A two-bond 31P scalar coupling is expected for [(SiO)Au(PMe3)2], but not for [(SiO)Au-PMe3], with a magnitude varying between 50 and 500 Hz. The INADEQUATE experiment was thus performed on {Au(PMe3)/SiO2}2 contacted with an excess of PMe3 since the 1D 31P CPMAS spectrum had the best signal to noise ratio. In the 1D refocused INADEQUATE spectrum (Fig. 3b), only one signal is observed at 7.7 ppm, which fully confirms its assignment to [(SiO)Au(PMe3)2]; the absence of a signal around −17 ppm confirmed its assignment to a mono-adduct surface species, [(SiO)Au-PMe3].
Overall, mono-phosphine adduct Au(I) species can be formed by grafting (Me3Si)2NAuPMe3 onto the isolated silanols of SiO2-(700), but the observation of two signals in a 2:1 ratio in the 31P NMR show that two [(SiO)Au-PMe3] species are in fact present and evidence their slightly different environments. Upon further reaction with additional PMe3, these species are either unreactive or transformed into [(SiO)Au(PMe3)2], respectively (Scheme 1b). Formation of such species was previously observed when [(SiO)Au–L] was similarly contacted with PMe3 (Scheme 1a). This corroborates the fact that both initial surface species [(SiO)Au-PMe3] or [(SiO)Au–L], obtained by grafting Me3PAuN(SiMe3)2 and [AuN(SiMe3)2]4, respectively, are indeed surrounded by similar yet heterogeneous environments (e.g. close proximity or not to adjacent surface siloxane bridges) as expected for an amorphous silica surface: some sites can accommodate only one PMe3 ligand suggesting the presence of nearby siloxane bridges probably coordinated to Au(I), while others have a much more open coordination sphere thus allowing the formation of bis-phosphine adducts (Scheme 2).
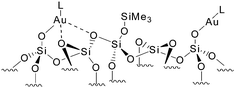 | ||
| Scheme 2 Au(I)-L surface species closely surrounded or not by surface siloxane bridges. | ||
In particular the results here show that the isolated surface silanols of silica partially dehydroxylated at 700 °C are surrounded by heterogeneous environments, limiting the possibility to access true “single-site” surface species, and it further illustrates previous findings by us and others where such heterogeneous environments of amorphous silica surfaces have been put forward to explain differences in structures, dynamics,14 and reactivities of silica supported surface organometallic complexes, e.g. Zr,15Ta,16Cr17 and Os.18 Further work is currently underway to better control the surface heterogeneity of oxide supports without losing the advantage of site isolation.19
Acknowledgements
DG and DL thank the Cluster de recherche Chimie de la Région Rhône-Alpes and the MIT foundation, respectively. This research was supported in part by ANR (ANR08-BL34507).Notes and references
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef CAS.
- T. J. Marks, Acc. Chem. Res., 1992, 25, 57–65 CrossRef CAS.
- J. Guzman and B. C. Gates, Dalton Trans., 2003, 3303–3318 RSC.
- K. L. Fujdala, R. L. Brutchey and T. D. Tilley, Topics in Organometallic Chemistry, 2005, 16, 69–115 Search PubMed.
- M. Tada and Y. Iwasawa, Coord. Chem. Rev., 2007, 251, 2702–2716 CrossRef CAS.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS.
- F. Blanc, R. Berthoud, A. Salameh, J. M. Basset, C. Copéret, R. Singh and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 8434–8435 CrossRef CAS.
- C. Copéret, Dalton Trans., 2007, 5498–5504 RSC.
- Y. I. Ermakov and B. N. Kuznetsov, Kinetika i Kataliz, 1977, 18, 1167–1178 CAS.
- D. Gajan, K. Guillois, P. Delichere, J. M. Basset, J. P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667–14669 CrossRef CAS.
- J. M. Basset, J. P. Candy and C. Copéret, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, pp. 499–553 Search PubMed.
- (a) S. Cadars, J. Sein, L. Duma, A. Lesage, T. N. Pham, J. H. Baltisberger, S. P. Brown and L. Emsley, J. Magn. Reson., 2007, 188, 24–34 CrossRef CAS; (b) F. Fayon, G. Le Saout, L. Emsley and D. Massiot, Chem. Commun., 2002, 1702–1703 RSC; (c) A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- (a) L. Duma, W. C. Lai, M. Carravetta, L. Emsley, S. P. Brown and M. H. Levitt, ChemPhysChem, 2004, 5, 815–833 CrossRef CAS; (b) F. Fayon, D. Massiot, M. H. Levitt, J. J. Titman, D. H. Gregory, L. Duma, L. Emsley and S. P. Brown, J. Chem. Phys., 2005, 122, 194313 CrossRef; (c) M. M. Maricq and J. S. Waugh, J. Chem. Phys., 1979, 70, 3300–3316 CrossRef CAS.
- J. Gath, G. L. Hoatson, R. L. Vold, R. Berthoud, C. Copéret, M. Grellier, S. Sabo-Etienne, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2009, 11, 6962–6971 RSC.
- F. Rataboul, A. Baudouin, C. Thieuleux, L. Veyre, C. Copéret, J. Thivolle-Cazat, J. M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2004, 126, 12541–12550 CrossRef CAS.
- S. Soignier, M. Taoufik, E. Le Roux, G. Saggio, C. Dablemont, A. Baudouin, F. Lefebvre, A. de Mallmann, J. Thivolle-Cazat, J. M. Basset, G. Sunley and B. M. Maunders, Organometallics, 2006, 25, 1569–1577 CrossRef CAS.
- E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, Chem. Rev., 2005, 105, 115–183 CrossRef CAS.
- R. Berthoud, N. Rendon, F. Blanc, X. Solans-Monfort, C. Copéret and O. Eisenstein, Dalton Trans., 2009, 5879–5886 RSC.
- (a) T. K. Maishal, J. Alauzun, J. M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reye, L. Veyre and C. Thieuleux, Angew. Chem., Int. Ed., 2008, 47, 8654–8656 CrossRef CAS; (b) I. Karame, M. Boualleg, J. M. Camus, T. K. Maishal, J. Alauzun, J. M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reye, L. Veyre and C. Thieuleux, Chem.–Eur. J., 2009, 15, 11820–11823 CrossRef CAS; (c) A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Mieville, J. Alauzun, A. Roussey, C. Thieuleux, A. Medhi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization methods. See DOI: 10.1039/c0sc00579g |
| This journal is © The Royal Society of Chemistry 2011 |