Gold(I)-catalyzed intermolecular (4 + 2) cycloaddition of allenamides and acyclic dienes†
Hélio
Faustino
a,
Fernando
López
*b,
Luis
Castedo
a and
José L.
Mascareñas
*a
aDepartamento de Química Orgánica, Centro Singular de Investigación en Química Biológica y Materiales Moleculares, y Unidad Asociada al CSIC. Universidad de Santiago de Compostela, 15782, Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es; Fax: +34 981 595012; Tel: +34 881814405
bInstituto de Química Orgánica General, CSIC, Juan de la Cierva 3, 28006, Madrid, Spain. E-mail: fernando.lopez@iqog.csic.es; Fax: +34 915644853; Tel: +34 915622900
First published on 14th February 2011
Abstract
A new type of intermolecular (4 + 2) cycloaddition, based on a gold-catalyzed reaction between allenamides and acyclic conjugated dienes, is reported. The annulation, which fails under standard Diels–Alder conditions, provides a straight entry to a variety of differently substituted cyclohexenes, and takes place with excellent regio- and diastereoselectivity.
Intermolecular (4 + 2) Diels–Alder cycloadditions are among the most powerful synthetic transformations so far described;1 however, their effectiveness is usually restricted to the use of properly biased dienes and dienophiles. The development of methods that allow us to perform (4 + 2) cycloaddition of Diels–Alder-inactive substrates would significantly expand the scope and potential of this ring assembly strategy.
We and others have recently described gold-catalyzed intramolecular (4 + 2) cycloadditions of allene-tethered-dienes,2 reactions that efficiently afford trans-fused bicyclic systems under very mild conditions. Conversely, more challenging, intermolecular versions of these cycloadditions have remained elusive. Herein, we report the discovery and implementation of such intermolecular process, namely a highly selective gold-catalyzed (4 + 2) cycloaddition between non-activated 1,3-dienes and allenamides, a particularly accessible and versatile type of allenic scaffold.3 To the best of our knowledge, this methodology constitutes the first transition metal-catalyzed intermolecular (4 + 2) cycloaddition between allenes (2C) and 1,3-dienes (4C),4 as well as one of the very few types of intermolecular cycloadditions of allenes promoted by gold or platinum catalysts.5,6
A preliminary reactivity screening using isoprene (2a) revealed that its treatment with electronically neutral allenes, such as 3-methylbuta-1,2-diene (1a, Fig. 1), in the presence of different sources of Pt or Au catalysts, leads to intractable mixtures of products. These results confirmed that the development of intermolecular variants of this type of annulations between allenes and dienes is not straightforward.7 Electronically rich allenes such as 1b or 1c also led to complex reaction mixtures.8
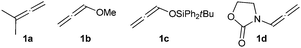 | ||
| Fig. 1 | ||
Gratifyingly, treatment of isoprene with the allenamide 1d in the presence of PPh3AuCl/AgSbF6, at −15 °C, produced the (4 + 2) cycloadduct 3da in 33% yield together with the (2 + 2) cycloadduct 4da, which was isolated in 19% yield. (Table 1, entry 1).9 As shown in entries 1–4, this reaction could also be promoted by several other cationic Au-catalysts (B-D). In all the cases, in addition to the (4 + 2) adduct, we also isolated variable amounts of the (2 + 2) cyclobutane derivative 4da (14–20% yield).10 Remarkably, neutral gold chloride catalysts turned out to be less active but more selective in favor of the (4 + 2) process (Table 1, entries 5–7), with AuCl being the most effective as we observed only traces of the side (2 + 2) adduct (Table 1, entry 7). Thus, treatment of 1d with 5 mol% of AuCl in presence of isoprene (6 equiv) at rt afforded, in a completely regio- and highly stereoselective manner, the (4 + 2) cycloadduct 3da in a 65% isolated yield.
 .
.| Entry | Catalyst | Solvent | Time (h) | T (°C) | Conv (%)b | 3da (%)c | 4da (%)c |
|---|---|---|---|---|---|---|---|
a
Allene
1d (1 equiv) was added to a mixture of 2a (6 equiv) and catalyst (5 mol%), in the specified solvent (0.1 M).
b Conversion by 1H-NMR.
c Isolated yields. Adducts 3da and 4da are predominantly obtained as Z isomers (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E ratio > 9:1); determined by 2D-NMR.
d Z:E ratio = 93:7.
e 3 equiv of diene were used.
f 2 equiv of diene were used.
g
Pt:L ratio = 1:2.
h
Degradation of 1d was seen. :E ratio > 9:1); determined by 2D-NMR.
d Z:E ratio = 93:7.
e 3 equiv of diene were used.
f 2 equiv of diene were used.
g
Pt:L ratio = 1:2.
h
Degradation of 1d was seen. 
|
|||||||
| 1 | Ph3PAuCl/AgSbF6 (A) | CH2Cl2 | 0.5 | −15 | 100 | 33 | 19 |
| 2 | B | CH2Cl2 | 0.1 | −15 | 100 | 41 | 16 |
| 3 | C | CH2Cl2 | 0.1 | −15 | 100 | 47 | 20 |
| 4 | D | CH2Cl2 | 6 | −15 | 100 | 64 | 14 |
| 5 | E | CH2Cl2 | 24 | rt | 100 | 46 | 6 |
| 6 | AuCl3 | CH2Cl2 | 48 | rt | 87 | 28 | 4 |
| 7 | AuCl | CH2Cl2 | 6 | rt | 100 | 65d | <2 |
| 8e | AuCl | CH2Cl2 | 6 | rt | 100 | 66d | <2 |
| 9f | AuCl | CH2Cl2 | 12 | rt | 100 | 55d | <2 |
| 105 | [PtCl2(C2H4)]2/P(otol)3g | EtOAc | 6 | rt | 10 | 2 | 0 |
| 11 | [PtCl2(C2H4)]2/P(otol)3g | CH2Cl2 | 5 | rt | 57 | 15 | 0 |
| 12 | PtCl2 | CH2Cl2 | 24 | rt | 0 | — | — |
| 13 | CSA | CH2Cl2 | 5 | rt | —h | — | — |
| 14 | PPTS | CH2Cl2 | 5 | rt | —h | — | — |
| 15 | AgSbF6 | CH2Cl2 | 18 | 40 | 0 | — | — |
| 16 | — | THF | 4 | 65 | 0 | — | — |
| 17 | — | toluene | 18 | 150 | 0 | — | — |
Importantly, reducing the amount of isoprene from six to three equivalents did not influence the efficiency of the process (Table 1, entry 8), whereas the use of just 2 equivalents provided a slightly lower reaction yield (Table 1, entry 9). Remarkably, Pt catalysts that had been previously found useful for inducing intermolecular (3 + 2) cycloadditions5 gave very poor results in this process (Table 1, entries 10–11). On the other hand, control experiments with Brønsted acids or silver salts revealed that neither of them are able to promote the annulation (Table 1, entries 13–15), which, as expected, is also unfeasible under thermal conditions (Table 1, entries 16–17). This result confirms the Au-catalyzed nature of the process, as opposed to the recent thermal intramolecular cycloadditions of furan-tethered allenamides.9g
Once established an optimum catalytic system, we evaluated the versatility and scope of the process. As shown in Table 2, allenamide 1d undergoes the cycloaddition reaction with a variety of differently substituted acyclic dienes (2b-2k). Thus, reaction of 1d with 2,3-dimethylbuta-1,3-diene (2b) proceeded in just 10 min to provide a 76% of the expected cycloadduct 3db, which was isolated as a 93:7 mixture of Z:E isomers (Table 2, entry 1). Reducing the amount of diene from three to two equivalents slowed the reaction, but 3db could still be isolated in a satisfactory 63% yield (entry 2). Conjugated dienes 2c and 2d, equipped with a methyl group at their external positions, also reacted efficiently, providing the corresponding cycloadducts 3dc and 3dd in a completely regio- and highly diastereoselective manner (Table 2, entries 3 and 4). 2,4-Hexadiene 2e, with methyl groups at the distal positions, afforded a single (4 + 2) isomer 3de in 44% yield (67% conversion, Table 2, entry 5). Curiously, a better yield (67%) was obtained in this case when the reaction was carried out at −15 °C with the more reactive cationic catalyst B, which features a N-heterocyclic carbene ligand (Table 2, entry 6).11 Complex B was certainly the catalyst of choice for the reaction of allenamide 1d with 1,4-disubstituted diene 2f, reaction that gave cycloadduct 3df as the only identifiable adduct in 52% yield (Table 2, entries 7 and 8). Diene 2g, with two exomethylene groups, also participated in the cycloaddition process affording, upon treatment with AuCl (5 mol%) for 2 min, the expected adducts 3dg and 3dg′ in a good 80% global yield (Table 2, entry 9).
 .a
.a
| Entry | 1 | Diene (2) | (4 + 2) adduct (3) | [Au] | Time | 3 (%)b |
|---|---|---|---|---|---|---|
|
a
Allene
1 (1 equiv) was added to a mixture of 2 (3 equiv) and catalyst (5 mol%), in CH2Cl2 (0.1 M) at rt, unless otherwise noted; >99% conversions (1H-NMR).
b Isolated yields of 3 (Z:E ratio > 93%).
c 2 equiv of diene were used.
d 6 equiv of diene (unoptimized) were used.
e 10 mol% of catalyst.
f Diene 2d consists of a 7:3 mixture of Z:E isomers.
g 67% conversion.
h Reaction carried out at −15 °C.
i The corresponding (2 + 2) adduct 4de was also isolated in 17% yield;8 Global combined yield: 84%.
j Ratio of regioisomers 3dg:3dg′ = 1.8:1.
k A side adduct (5dh) was also isolated (13%).12 Global combined yield: 89%.
l Conversion (by 1H-NMR).
|
||||||
| 1 | 1d |

|

|
AuCl | 10 min | 76 |
| 2c | 1d | AuCl | 6 h | 63 | ||
| 3d,e | 1d |

|
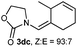
|
AuCl | 1 h | 60 |
| 4d,f | 1d |

|
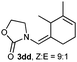
|
AuCl | 6 h | 62 |
| 5d | 1d |

|

|
AuCl | 20 h | 44g |
| 6h | 1d | B | 25 min | 67i | ||
| 7 | 1d |

|

|
AuCl | 7 h | 15 |
| 8h | 1d | B | 5 min | 52 | ||
| 9 | 1d |

|

|
AuCl | 2 min | 80j |
| 10 | 1d |
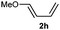
|

|
AuCl | 2 min | 76k |
| 11 | 1d |

|
|
AuCl | 10 min | 96 |
| 12c | 1d | AuCl | 4 h | 88 | ||
| 13 | 1d |

|

|
AuCl | 26 h | 81e |
| 14c | 1d |

|
|
AuCl | 15 h | 70 |
| 15 | 1e |
|

|
AuCl | 72h | 63l |
| 16h | 1e | B | 1h | 94 | ||
| 17 | 1f |

|

|
AuCl | 20 min | 79 |
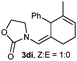
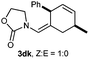
Donor substituents at the diene were well tolerated. Indeed, the reaction of allenamide 1d with diene 2h gave the corresponding adduct as a single regio- and diastereoisomer, in 76% yield (Table 2, entry 10). Similarly, the presence of aryl substituents at the diene provided very clean and selective reactions, so the desired (4 + 2) adducts were obtained with complete selectivities and good yields (Table 2, entries 11–14), even when only two equivalents of diene were employed (Table 2, entries 12, 14).
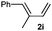
Also importantly, the reaction is not limited to allenamide 1d; other allenamides, such as 1e, or the chiral disubstituted allenamide 1f, equipped with a methyl group at the distal position, also participated in the process, affording the corresponding adducts 3ei and 3fi in good or excellent yields, and as single regio and diastereoisomers (Table 2, entries 15 – 17).
Confirmation of the structure and stereochemical assignment of all the products was unambiguously achieved by NMR analysis and in many cases, such as for cycloadducts 3db, 3dd, 3de, 3df, 3dj, 3di and 3fi, we could also resolve their structure by X-ray diffraction analysis (Fig. 2).8
 | ||
| Fig. 2 X-Ray structures of adducts 3de, 3fi.8 | ||
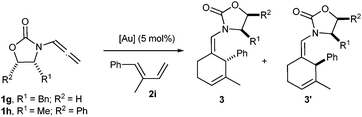
The presence of an oxazolidinone moiety in the allenamide unit brings excellent opportunities for the development of asymmetric variants of these cycloadditions. As a first approach toward this end, we prepare chiral allenamides 1g and 1h, easily accessible in two simple steps from commercially available oxazolidinone precursors.8,9a,c The AuCl-catalyzed cycloaddition of 1g with 2i led to a 5: 1 mixture of diastereoisomers 3gi and 3gi′, which were isolated in an excellent 95% combined yield (Table 3, entry 1).13 The diastereoselectivity of the process could not be improved by lowering the reaction temperature to 0 °C (Table 3, entry 2); however, by using the more reactive catalyst B, we could perform the reaction at −15 °C, obtaining an excellent 17:1 diastereoisomeric ratio and an overall 95% yield (Table 3, entry 3).14
 .a
.a
| Entry | 1 | [Au] | T (°C) | Time | Ratio (3: 3′)b | Yield (%)c |
|---|---|---|---|---|---|---|
|
a
Allene
1 (1 equiv) was added to a mixture of 2i (3 equiv) and catalyst (5 mol%), in CH2Cl2 (0.1 M) either at rt (AuCl), or −15 °C (catalyst B); > 99% conversions.
b
dr determined by 1H-NMR on the crude mixtures.
c Isolated yields of the mixture of both isomers (Z:E ratio = 1:0).
d 10 mol% of catalyst was used.
|
||||||
| 1 | 1g | AuCl | rt | 2 h | 5: 1 (3gi: 3gi′) | 95 |
| 2 | 1g | AuCl | 0 | 3.5 h | 5: 1 (3gi: 3gi′) | 62 |
| 3 | 1g | B | −15 | 10 min | 17: 1 (3gi: 3gi′) | 95 |
| 4d | 1h | AuCl | rt | 10 min | 1: 0 (3hi: 3hi′) | 99 |
| 5 | 1h | B | −15 | 10 min | 1: 0 (3hi: 3hi′) | 99 |
On the other hand, cycloadditions of chiral allenamide 1h with diene 2i were completely selective and very efficient with both catalysts. Indeed, under AuCl catalysis (10 mol%), a single diastereoisomer 3hi was obtained in 99% yield (Table 3, entry 4), whereas employing catalyst B, 3hi could be obtained in identical yield after just 10 min at −15 °C (Table 3, entry 5). The stereochemical assignment of 3hi as well as that of 3gi could be successfully established by X-ray analysis (Fig. 3).8
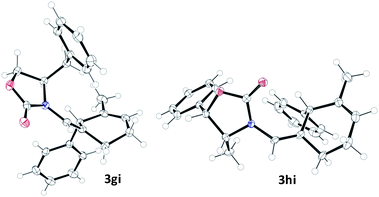 | ||
| Fig. 3 X-Ray structures of adducts 3gi and 3hi. | ||
The exo-enamide group present in the (4 + 2) cycloadducts of type 3 should provide for divergent synthetic elaborations.15 As a preliminary test we found that treatment of adduct 3hi under acidic conditions followed by reduction with NaBH4 yielded the expected alcohol in 71% yield (dr: 9:1, Scheme 1).
 | ||
| Scheme 1 | ||
From a mechanistic point of view, the regioselectivity of the process and the formation of secondary minor (2 + 2) cycloadducts suggest that, at least in some cases, the reaction might proceed through an stepwise cationic pathway such as that shown in Scheme 2. Thus, activation of the allene by the Au catalyst would afford a Au-allyl cation species of type I.2a,b,e Nucleophilic intermolecular interception of I by the diene would provide a second cationic intermediate II. This would be the regioselectivity-determining step, with the formation of the more substituted allyl cation II being favored. Finally, a ring closing process through attack of the enamide group to the more accessible α-carbon of the allyl cation, and elimination of the Au complex would yield the final (4 + 2) adduct 3. Alternatively, cyclobutane derivatives of type 4 would be produced by the attack of the enamide group at the internal γ-carbon of the allyl cationic species II.
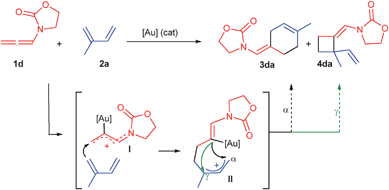 | ||
| Scheme 2 One potential mechanistic pathway for the Au-catalyzed cycloaddition of allenamides 1 and conjugated dienes | ||
In consonance with this mechanism, the Au-catalyzed reaction of 1d and 2b in the presence of exogenous MeOH (3 equiv) provided, besides 3db (40% yield), the allyl methyl ethers 7db and 8db in a global 14% yield (Scheme 3). These compounds must result from the nucleophilic trapping of intermediate II at its α and γ positions. Unfortunately, the yield of these compounds could not be further improved and performing the same reaction with other dienes such as 2a or 2i, instead of 2b, did not allow to isolate analogue compounds of type 7 or 8. Therefore, other mechanisms, in particular those involving either a direct concerted (4 + 2) cycloaddition of I with the diene, or a (4 + 3) cycloaddition followed by ring contraction,2 could also be operative. The predominance of either pathway might depend on both, the structure of the diene and the nature of the catalyst.
 | ||
| Scheme 3 (4 + 2) cycloaddition in the presence of MeOH (3 equiv). | ||
In conclusion, we have developed a new type of (4 + 2) cycloaddition reaction, namely a gold-catalyzed annulation of allenamides and acyclic conjugated dienes. The method provides synthetically appealing cyclohexene derivatives in a highly or completely selective manner and shows a wide scope and generality. Work to develop catalytic, enantioselective variants and to gain a deeper mechanistic understanding is underway.
Acknowledgements
This work was supported by the Spanish MEC [SAF2007-61015, SAF2010-20822-C02 and Consolider-Ingenio 2010 (CSD2007-00006)], CSIC and Xunta de Galicia (GRC2010/12, INCITE09 209 122 PR). HF acknowledges Fundação para a Ciência e a Tecnologia–Portugal for a PhD Grant SFRH/BD/60214/2009.Notes and references
- (a) F. Fringuelli and A. Taticchi, The Diels- Alder Reaction: Selected Practical Methods; Wiley: Chichester, U.K., 2002 Search PubMed; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS.
- (a) P. Mauleón, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348 CrossRef CAS; (b) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020 CrossRef CAS; (c) D. Benitez, E. Tkatchouk, A. Z. González, W. A. Goddard and F. D. Toste, Org. Lett., 2009, 11, 4798 CrossRef CAS; (d) A. Z. González and F. D. Toste, Org. Lett., 2010, 12, 200 CrossRef; (e) M. Alcarazo, T. Stork, A. Anoop, W. Thiel and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 2542 CrossRef CAS.
- For a review on the synthetic utility of allenamides, see: (a) L.-L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 CrossRef. For other types of Au-catalyzed reactions of allenamides, see: (b) A. W. Hill, M. R. J. Elsegood and M. C. Kimber, J. Org. Chem., 2010, 75, 5406 CrossRef CAS; (c) M. C. Kimber, Org. Lett., 2010, 12, 1128 CrossRef CAS.
- For a Rh-catalyzed intermolecular cycloaddition between unactivated vinylallenes (4C) and alkynes (2C), see: (a) M. Murakami, M. Ubukata, K. Itami and Y. Ito, Angew. Chem., Int. Ed., 1998, 37, 2248 CrossRef CAS.
- We are only aware of a Pt-catalyzed (3 + 2) cycloaddition of allenyl silyl ethers and enol ethers, see: H. Kusama, M. Ebisawa, H. Funami and N. Iwasawa, J. Am. Chem. Soc., 2009, 131, 16352 Search PubMed.
- For recent reviews on Au- and Pt-catalyzed reactions, see: (a) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS; (c) E. Jiménez-Nuñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef CAS; (d) H. C. Shen, Tetrahedron, 2008, 64, 7847 CrossRef CAS.
- Allenes and dienes may react in several ways in the presence of Au catalysts. For instance, we have shown that allenes can behave as 2C or 3C atom partners in their gold-catalyzed intramolecular cycloadditions with dienes, see ref. 2 and: (a) B. Trillo, F. López, M. Gulías, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2008, 47, 951 CrossRef CAS; (b) B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, Chem.–Eur. J., 2009, 15, 3336 CrossRef CAS. On the other hand, Au catalysts might also activate 1,3-dienes: (c) F. López and J. L. Mascareñas, Chem.–Eur. J., 2011, 17, 418 CrossRef CAS; (d) C. Brouwer and C. He, Angew. Chem., Int. Ed., 2006, 45, 1744 CrossRef CAS; (e) R. V. Nguyen, X. Q. Yao and C. J. Li, Org. Lett., 2006, 8, 2397 CrossRef CAS.
- See the Supplementary Information† for more details.
- It has been shown that allenamides can participate in uncatalyzed hetero-(4 + 2) inverse electron demand Diels–Alder cycloadditions, see: (a) L.-L. Wei, H. Xiong, C. J. Douglas and R. P. Hsung, Tetrahedron Lett., 1999, 40, 6903 CrossRef CAS; (b) L.-L. Wei, R. P. Hsung, H. Xiong, J. A. Mulder and N. T. Nkansah, Org. Lett., 1999, 1, 2145 CrossRef CAS; (c) C. R. Berry and R. P. Hsung, Tetrahedron, 2004, 60, 7629 CrossRef CAS; (d) M. Kimura, Y. Wakamiya, Y. Horino and Y. Tamaru, Tetrahedron Lett., 1997, 38, 3963 CrossRef CAS; (e) Y. Horino, M. Kimura, S. Tanak, T. Okajima and Y. Tamaru, Chem.–Eur. J., 2003, 9, 2419 CrossRef CAS. Two specific examples of normal electron demand (4 + 2) reactions have also been reported, see: (f) J. P. Bacci, K. L. Greenman and D. L. Van Vranken, J. Org. Chem., 2003, 68, 4955 CrossRef CAS; (g) A. G. Lohse and R. P. Hsung, Org. Lett., 2009, 11, 3430 CrossRef CAS.
- For other types of (2 + 2) cycloadditions employing different allenamides, see: (a) M. Kimura, Y. Horino, Y. Wakamiya, T. Okayima and Y. Tamaru, J. Am. Chem. Soc., 1997, 119, 10869 CrossRef CAS. For Au-catalyzed intramolecular (2 + 2) cycloadditions of allene-tethered alkenes, see ref. 2e and: (b) M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402 CrossRef CAS; (c) H. Teller, S. Flugge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949 CAS. For a recent review on (2 + 2) cycloadditions of allenes, see: (d) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC. For a recent Au-catalyzed intermolecular (2 + 2) cycloaddition between alkynes and alkenes, see: (e) V. López-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292–9294 CrossRef CAS.
- Additionally, in this reaction we also isolated a minor amount (17%) of the corresponding (2 + 2) adduct 4de.
 .
. - Submitting the (4 + 2) adduct 3dh to the reaction conditions provides 5dh, which confirms that 5dh is formed in situ from 3dh.
 .
. - Identification of isomeric adducts 3gi and 3gi′ could be performed by NMR analysis. Moreover, the major isomer, 3gi, could be purified by crystallography, and further identified by X-ray diffraction8.
- The (2 + 2) adduct (4gi) was not detected.
- For different reactions on related enamides, see: Diels–Alder, (a) F. Gallier, H. Hussain, A. Martel, A. Kirschning and G. Dujardin, Org. Lett., 2009, 11, 3060 CrossRef CAS; Epoxidation: (b) W. Adam, S. G. Bosio, N. J. Turro and B. T. Wolff, Org. Lett., 2003, 5, 819 CrossRef CAS; (c) W. Adam, S. G. Bosio and B. T. Wolff, J. Org. Chem., 2004, 69, 1704 CrossRef CAS; (d) Photooxygenation: W. Adam, S. G. Bosio and N. J. Turro, J. Am. Chem. Soc., 2002, 124, 8814 Search PubMed; Halogenation: (e) C. Ko, R. P. Hsung, Z. F. Al-Rashid, J. B. Feltenberger, T. Lu, H. J-Yang, Y. Wei and C. A. Zificsak, Org. Lett., 2007, 9, 4459 CrossRef CAS; Hydrogenation: (f) B. Gourdet, M. E. Rudkin and H. W. Lam, Org. Lett., 2010, 12, 2554 CrossRef CAS; (g) Asymmetric dihydroxylation: B. Gourdet and H. W. Lam, Angew. Chem., Int. Ed., 2010, 49, 8733 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, crystallographic data. CCDC reference numbers 804843–804850. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00630k |
| This journal is © The Royal Society of Chemistry 2011 |