DOI:
10.1039/C0SC00574F
(Edge Article)
Chem. Sci., 2011,
2, 625-632
DNA-templated combinatorial assembly of small molecule fragments amenable to selection/amplification cycles†
Received
16th November 2010
, Accepted 4th January 2011
First published on 24th January 2011
Abstract
The discovery of small molecule probes which selectively modulate biological pathways is a cornerstone in the development of new therapeutics. Progress in our ability to access libraries of biologically relevant small molecules in conjunction with streamlined screening technologies have also enabled a more systematic approach to chemical biology. Nevertheless, the current state of the art still requires a large infrastructure and only a small fraction of the proteome has been addressed thus far. The emergence of technologies based on nucleic acid encoding of small molecules presents a new screening paradigm. We describe a method based on DNA-templated combinatorial display of PNA-encoded drug fragments affording 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 combinations which can be amplified following a selection. This concept was demonstrated with a screen against a representative target, carbonic anhydrase, by iterative cycles of affinity selection, amplification of DNA template and “translation” back into selected library members. The results show a clear convergence towards combinations which, upon resynthesis as covalent adducts, proved to bind cooperatively to carbonic anhydrase.
500 combinations which can be amplified following a selection. This concept was demonstrated with a screen against a representative target, carbonic anhydrase, by iterative cycles of affinity selection, amplification of DNA template and “translation” back into selected library members. The results show a clear convergence towards combinations which, upon resynthesis as covalent adducts, proved to bind cooperatively to carbonic anhydrase.
Introduction
The diversity space covered by screening technologies leveraged on iterative cycles of selection/amplification such as phage display1 and SELEX2 dwarf current small molecule screening technologies.3 An implicit requirement to harness the power of these biochemical screens at the small molecule level is an encoding technology which can be used to amplify selected library members. Several groups have reported technologies to prepare DNA-encoded libraries.4 Liu and co-workers have shown that DNA tags can be “translated” into small molecules using DNA-templated synthesis5 and have demonstrated in vitro selection of a pilot library of 65 macrocycles.6 More recently, they have developed and validated a codon system enabling larger libraries and prepared over 13000 peptide-based macrocycles,7 which led to the identification of new kinase inhibitors.8 Researchers at Vipergen have reported a similar approach based on three-dimensional DNA architectures.9 Harbury and co-workers have exploited the selectivity of hybridization to route large mixtures of DNA tags through a combinatorial synthesis10–12 which has been applied to the synthesis of a peptoid library containing over 100 million putative compounds.13 Alternatively, two groups have reported the use of DNA-polymerase or DNA-ligase to extend a DNA tag after each step in a combinatorial synthesis.14–16 Upon selection of a library member against a target, the best binders are typically identified by amplification of the DNA tag and sequencing or microarray hybridization.17 Neri and coworkers have also shown that a library of small molecules can be hybridized to a complementary strand derivatized with a given pharmacophore to improve its affinity in a process akin to antibody maturation.18–20 To date, only Harbury and coworkers have demonstrated the “translation” of enriched sequences isolated from a selection back into a small molecule library for iterative screening cycles.13 We have been developing technologies to encode chemical libraries with peptide nucleic acids (PNAs)21 based on the premise that PNA is more robust than DNA and its chemistry more permissive for the preparation of small molecule libraries.22–25 However, a limitation of PNA relative to DNA is that it cannot be readily amplified. Inspired by the development of fragment-based approach to drug discovery,26–31 we reasoned that libraries of PNA-encoded fragments could be combinatorially assembled by hybridization to a complementary DNA library (Fig. 1). While this strategy is conceptually related to the work reported by Neri18–20 and coworkers, this architecture would provide a simple means of combining multiple libraries thus providing a combinatorial output of diversity. This strategy also lends itself to a rapid amplification of a selected subset of the library by rehybridization of the fragments after a selection, which is not possible with any previously reported technology.
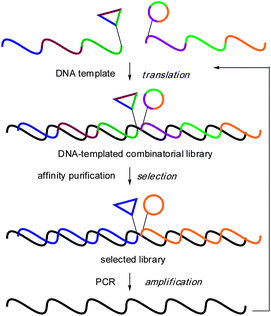 |
| | Fig. 1 DNA-templated combinatorial assembly, selection and amplification. | |
Results and discussion
Library synthesis
For fragments to be presented close in space upon combinatorial assembly onto a DNA template, two PNA libraries that hybridize contiguously are required, one bearing the small molecule at the N-terminus and the other at the C-terminus of the PNA. The use of two different libraries also ensures a dissymmetry in the combinatorial assembly and avoids redundant pairs (A1B1vs. B1A1). Based on the availability in our laboratory of microarrays containing 625 sequences complementary to a validated PNA codon system with homogeneous Tms,25 we opted to use this codon set and divide it into two libraries of 125 + 500 which would yield 62500 combinatorial pairs. To implement this strategy, we opted to start with biologically validated fragments32 (FDA approved drugs, bioactive natural products or fragments thereof) for the N-terminus library and developed chemistry yielding heterocyclic libraries with precedents as versatile pharmacophores for the C-terminus library.
Synthesis of heterocyclic libraries appended at the C-terminus of the PNA
We reasoned that libraries of heterocyclic pharmacophores (1,2,3-triazole 1, pyrazole 2 and pyrimidine 3) could arise from a common intermediate 5via divergent combinatorial pathways33,34 as shown in Fig. 2. Thus, a resin derivatized with a bifunctional linker (lysine) bearing on one side an azide-protected amino acid 5 could either be elaborated through cycloaddition reactions with alkynes or alternatively coupled to flavones to obtain intermediate 4 which would in turn be diversified with hydrazines or amidines. The synthesis was carried out on a rink resin as shown in Fig. 3. Intermediate 6 was coupled with 25 Fmoc amino acids and the amine group was converted to an azide to obtain intermediate 5 (see supplementary information† for the synthons and detailed synthetic procedures as well as intermediate characterization). Each pool was then encoded with the appropriate PNA codons through Fmoc-based PNA synthesis35 and the resins 8 were mixed and split into 20 new pools for cycloaddition with diverse alkyne synthons followed by PNA encoding. The different pools were mixed and cleaved from the resin (TFA) with concomitant deprotection of all side chains and nucleotides to obtain library 1. Alternatively, a subset of five pools of intermediate 5 were diverted through a complementary reaction pathway involving PNA encoding, mix and split of the resins followed by coupling to flavones and addition of the corresponding PNA codon to obtain five pools of intermediate 4. The resins were subjected to a third round of mix and split and treated with different hydrazines or amidines to obtain after PNA encoding and cleavage from the resin libraries 2 and 3 respectively. All synthons of diversity used in the library were commercially available and individually validated. After the completion of each reaction cycle (diversification, encoding), an analytical aliquot of resin was cleaved for quality control (MALDI analysis). All the chemistry was performed in an automated fashion using an Intavis Peptipep RS instrument on μmol scale which provides sufficient product for over 1000 screens. Automation of this process coupled to the use of commercially available synthons made it possible to access such libraries within a few weeks.
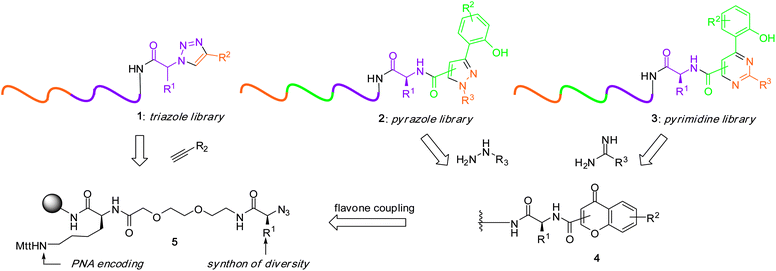 |
| | Fig. 2 Divergent pathways for PNA-encoded heterocyclic libraries. | |
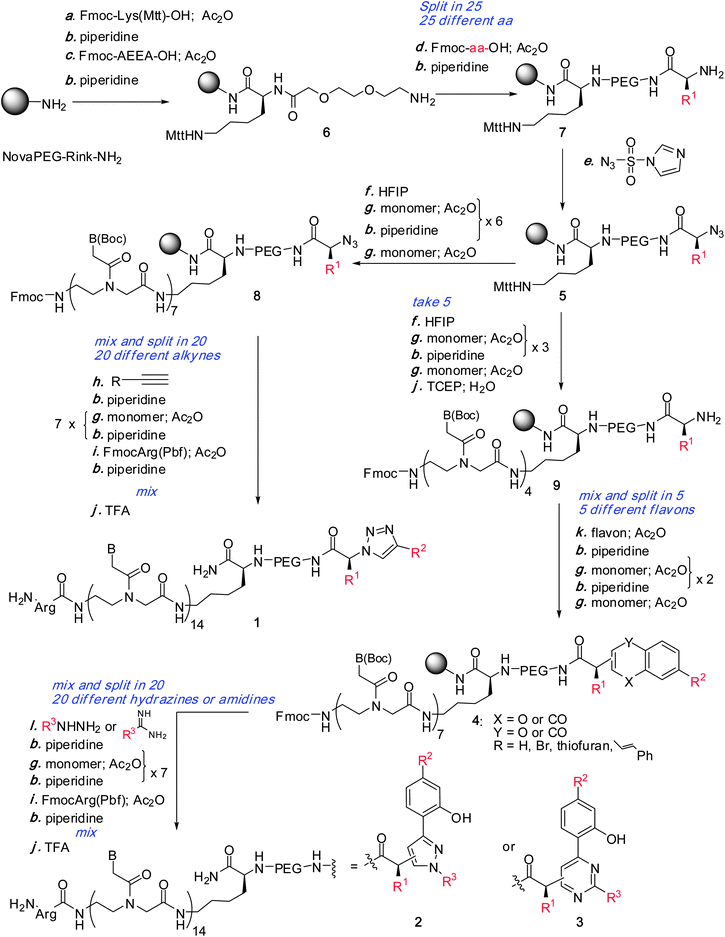 |
| | Fig. 3 Diversity-oriented synthesis of heterocyclic libraries 1, 2, 3 appended at the C-terminus of the PNA. | |
Synthesis of validated bioactive fragments conjugated at the N-terminus
A library of 125 PNAs was prepared by automated parallel synthesis on 2 μmol scale and derivatized with a short polyethylene (PEG) spacer to obtain resins 10 (Fig. 4). Four strategies were developed to link commercially available bioactive fragments to the PNAs (see Fig. 5 for selected examples of fragments). Fragments bearing a carboxyl group were simply activated (HOBt/DIC) and added to resins 10. To conjugate molecules bearing an amino or hydroxyl group, the N-terminus amine was converted into an activated carbamate (p-nitrophenylchloroformate) or an active ester (diglycolic anhydride; BOP/HOBt, DIPEA) and treated with the fragment in the presence of base. To access sulfonamides, the amino group was simply coupled to sulfonyl chlorides. The success of each conjugation was verified by MALDI of the cleavage products.
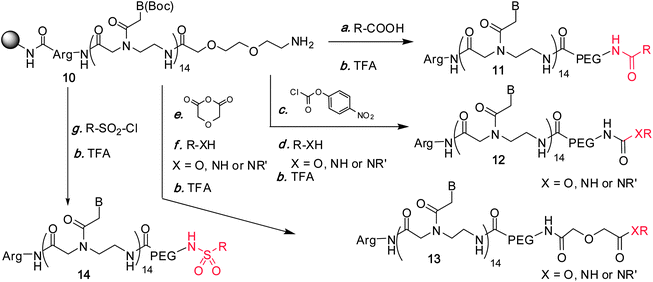 |
| | Fig. 4
Conjugation of bioactive pharmacophores to the N-terminus of PNA. | |
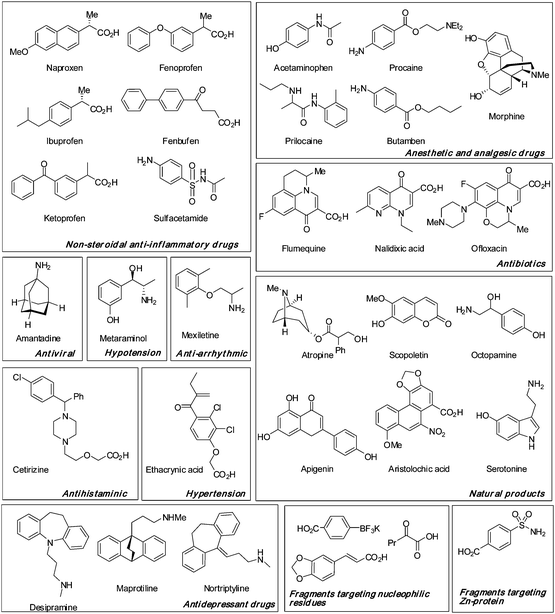 |
| | Fig. 5 Representative examples of biologically validated fragments conjugated to the N-terminus of the PNA. An aryl sulfonamide fragment (lower right corner) known to interact with the zinc atom of carbonic anhydrase was included. | |
DNA-templated combinatorial assembly of fragment libraries and screening
Considering that each PNA library (N-ter library and C-ter library) has a 14-mer tag,25 a 28-mer DNA template is required for the self assembly of the libraries. Preliminary experiments with different primer lengths flanking the DNA-template suggested that 20-mer primers afforded optimal amplification by PCR. The library of DNA templates containing two 20-mer primer flanking the encoding region (28-mer) was prepared by mix and split DNA synthesis and purified by PAGE to remove any truncated sequences. The DNA-templated combinatorial library was obtained by mixing the DNA library with an equimolar ratio of the two PNA libraries and complementary sequences to the primers to obtain a final library concentration of 600 nM. The mixture was heat denatured (10 min at 95 °C) and then allowed to cool to room temperature over one hour.
Library screening
To assess the potential of an iterative screening strategy, we opted to use carbonic anhydrase as a target. It has previously been used to validate several fragment-based screening approaches18,27,36 as well as a DNA-encoded library,6 and has a known selectivity for an aromatic sulfonamide fragment which was included in our library and would thus enable us to validate the efficiency of the selection. We envisioned performing iterative selection as illustrated in Fig. 6. Selection of the DNA-templated combinatorial library against an immobilized target should lead to an enrichment of productive combinations of fragments which could be recovered using a final stringent wash. As the instructions for the selected combinations are DNA-based, these instructions can be amplified by PCR. In order to translate the instructions back into a small molecule library, we reasoned that the PCR product could be immobilized, converted to a single strand and exposed to an excess of the fragment libraries in order to capture and reassemble the productive fragment combinations. This enriched library can be used for further rounds of selection. Thus, while we had previously demonstrated that PNA-encoded libraries could be selected against a given target,22,23,25 a significant extension of the present strategy is to capitalize on the programmable combinatorial assembly37,38 of PNA-tagged fragments onto a library of DNA templates to achieve a broader diversity. This approach effectively provides an encoding strategy for dynamic combinatorial libraries.27,28,30,31
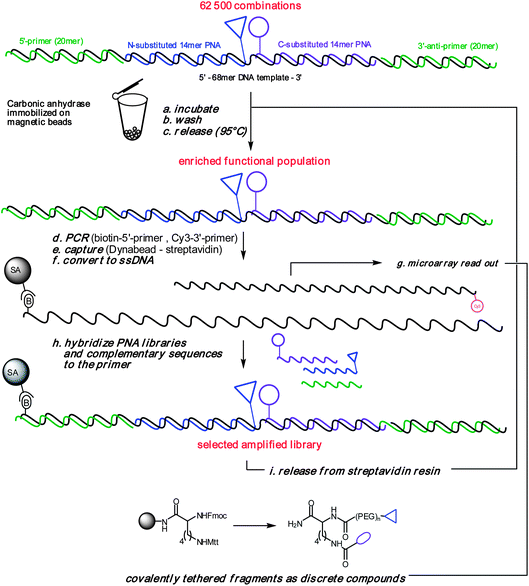 |
| | Fig. 6 General scheme for library selection/amplification. | |
The screen was carried out with 300 fmol of protein which corresponds to the minimum quantity of magnetic beads (10 μl) that can be comfortably isolated with a magnetic stand and 6 pmol of the library (10 μl). After 30 min incubation, the beads were pelleted with a magnetic stand, the supernatant removed, and the beads were washed 10 times with buffer. The DNA instructions corresponding to selected library members were recovered by heat denaturation and amplified by PCR with the primer for the templating strand bearing a biotin and the primer for the complementary strand bearing a fluorophore (Cy3). It is noteworthy that only a small fraction (0.02%) of the recovered solution from the selection was necessary for the PCR reaction suggesting the selection could be further miniaturized. The product of the PCR reaction was captured on a streptavidin resin and converted to single strand DNA to recover the Cy3-labeled strand.39 The PNA-encoded fragment libraries were hybridized to the immobilized strand for 30 min at 50 °C. The DNA-templated combinatorial library thus obtained was then released from the resin by heating in a solution of 5 mM biotin40 and subjected to three more rounds of selection using the same procedure. The evolution of the selection was visualized by hybridization of the Cy3 fluorescently labeled strand recovered from each round of selection (Fig. 7A). The same procedure was performed in parallel with bovine serum albumin (BSA) as a control.
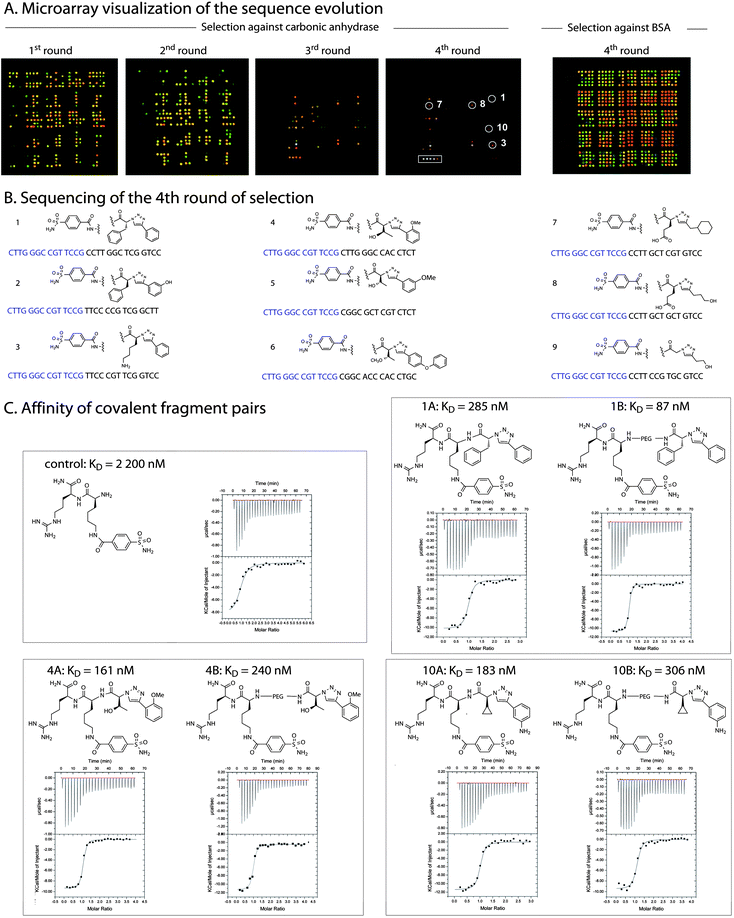 |
| | Fig. 7 Analysis of the selection and confirmation of selected fragments. A. Microarray analysis of each round of selection. The sequence corresponding to the sulfonamide fragment is shown as a box in the fourth round of selection against carbonic anhydrase, the sequence corresponding to the triazole fragments are shown as circles (the number corresponds to the sequencing number in B except for 10 which was not identified in the sequencing); B. Sequencing of nine random clones from the fourth round of selection. The structures of the fragments corresponding to each sequence are shown above the sequences; C. Affinity of covalent fragment pairs measured by isothermal titration calorimetry. | |
While there is a clear selection in the first round (Fig. 7 panel A), the library has not converged to a significant extent. After the fourth round, the brightest features on the array corresponds to the sequence encoding the aryl sulfonamide, a fragment known to bind carbonic anhydrase, and several triazole fragments. While the microarray readout provides a rapid assessment on the structure of selected fragments, it does not provide information regarding their pairing. It is interesting to note that the feature corresponding to the sulfonamide is significantly brighter than features corresponding to the triazole fragments suggesting that the nature of the substituent on the triazole is more permissive. To further establish the identity of the selected populations, the product of the fourth round of selection was cloned with a primer including the sequence of the sulfonamide fragment and sequenced to establish the identity of the selected fragment pairs (9 clones, sequences shown in Fig. 7B). Selection with the pyrazole (2) and pyrimidine (3) libraries did not afford similar convergence suggesting that the triazole may be a necessary feature in the interaction. Selection of the different libraries against bovine serum albumin (BSA), which is known for its promiscuity, failed to converge. A selection with the beads alone did not provide a significant amount of PCR product concurring the lack of affinity of any of the library members for the beads. The screen against carbonic anhydrase (4 rounds of selection) was carried out three times and consistently converged to the same fragments.
Selection for other target types (proteases, kinases or a carbohydrate binding protein) converged to different fragment combinations (data not shown) suggesting that the diversity space covered by the combinatorial assembly of these relatively small fragment sets are sufficiently diverse to address functionally distinct proteins.
The lack of a clear selection in the first round is not entirely surprising if one considers that the protein is in great excess (3000 fold) relative to each library member. This latter point is an intrinsic issue associated with screening large mixtures of compounds and underscores the advantage associated with the ability to amplify a selected population and translate it into a small molecule library for further rounds of selection. From an experimental standpoint, the selection, amplification and “translation” of the amplified DNA into the small molecule assembly only requires a few hours, thus enabling several rounds of selection to be performed within a day. Additionally, the cycle of amplification/“translation” does offer the possibility to reintroduce diversity in analogy to the natural evolution process.
Synthesis of covalent adducts from selected fragments
The convergence observed in the selection with carbonic anhydrase suggests that the selection/amplification approach presented herein is effective. However, the question remained whether the second fragment provided synergy41 in the binding to carbonic anhydrase. Three fragment pairs from the sequencing and microarray analysis (sequence 1, identified both by sequencing and microarray, sequence 4 identified by sequencing; 10 identified on the microarray – see Fig. 7 for numbering) were selected for resynthesis as covalent adducts. The chemistry was performed by solid phase synthesis using the same procedure as developed for the library preparation using both nitrogens of a lysine to connect the fragments. To vary the distance between the fragments, they were either connected directly to the lysine or with an additional 10Å polyethylene spacer (structures are shown in Fig. 7C). To ensure good water solubility and avoid artifacts in measuring their affinity by isothermal titration calorimetry (ITC) an arginine residue was added to each compound. As a control, the sulfonamide portion of the molecule lacking the second substituent was also prepared and measured to have an affinity of 2200 nM (Fig. 7C). The covalent adduct of the optimal fragment pair (87 nM, 1B, Fig. 7C) showed a 25-fold enhancement in affinity relative to the sulfonamide fragment itself. Interestingly, the same fragment pair with a shorter linker had a lower affinity of 285 nM suggesting that the shorter linker precludes the more favourable binding conformation. Other pairs of fragments had affinities ranging from 161 nM to 306 nM confirming a cooperative binding of the identified fragments in the selection.
Conclusions
Fragment-based drug design has gained tremendous momentum in the drug discovery process;42,43 however, the identification of low affinity fragments which bind cooperatively into a binding pocket remains experimentally challenging. The method reported herein provides a rapid means to do so with extremely small amounts of protein and library. From a chemistry standpoint, the libraries of heterocyclic pharmacophores further extend the scope of PNA-encoded synthesis. The different conjugation protocols developed enable the incorporation of diverse and validated fragment sets onto PNA tags.
The ability to perform multiple rounds of selection/amplification has been the key to the remarkable success of biochemical selection systems such as phage display and SELEX, to identify binders from peptide and nucleic acid libraries. The approach described herein extends the scope of iterative selection to small molecules broadly recognized as versatile pharmacophores.
Acknowledgements
This work was supported by a grant from the European Research Council (ERC 201749). The Institut Universitaire de France (IUF) is gratefully acknowledged for its support. Fellowships from the university of Strasbourg (JPD), the French Ministry of Research (MC) and MEC-Spain Xunta de Galicia (SA) are gratefully acknowledged.
Notes and references
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- T. Kodadek, Nat. Chem. Biol., 2010, 6, 162–165 CrossRef CAS.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CAS; J. Nielsen, S. Brenner and K. D. Janda, J. Am. Chem. Soc., 1993, 115, 9812–9813 CrossRef CAS For recent reviews, see: J. Scheuermann and D. Neri, ChemBioChem, 2010, 11, 931–937 Search PubMed; M. A. Clark, Curr. Opin. Chem. Biol., 2010, 14, 396–403 CrossRef CAS.
- Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961–6963 CrossRef CAS; X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS.
- B. N. Tse, T. M. Snyder, Y. Shen and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 15611–15626 CrossRef CAS.
- R. E. Kleiner, C. E. Dumelin, G. C. Tiu, K. Sakurai and D. R. Liu, J. Am. Chem. Soc., 2010, 132, 11779–11791 CrossRef CAS.
- M. H. Hansen, P. Blakskjaer, L. K. Petersen, T. H. Hansen, J. W. Hoejfeldt, K. V. Gothelf and N. J. V. Hansen, J. Am. Chem. Soc., 2009, 131, 1322–1327 CrossRef CAS.
- D. R. Halpin, J. A. Lee, S. J. Wrenn and P. B. Harbury, PLoS Biol., 2004, 2, e175 CrossRef.
- D. R. Halpin and P. B. Harbury, PLoS Biol., 2004, 2, e173 CrossRef.
- D. R. Halpin and P. B. Harbury, PLoS Biol., 2004, 2, e174 CrossRef.
- S. J. Wrenn, R. M. Weisinger, D. R. Halpin and P. B. Harbury, J. Am. Chem. Soc., 2007, 129, 13137–13143 CrossRef CAS.
- L. Mannocci, Y. X. Zhang, J. Scheuermann, M. Leimbacher, G. De Bellis, E. Rizzi, C. Dumelin, S. Melkko and D. Neri, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17670–17675 CrossRef CAS.
- F. Buller, Y. Zhang, J. Scheuermann, J. Schafer, P. Buhlmann and D. Neri, Chem. Biol., 2009, 16, 1075–1086 CrossRef CAS.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS.
- J. Scheuermann, C. E. Dumelin, S. Melkko, Y. X. Zhang, L. Mannocci, M. Jaggi, J. Sobek and D. Neri, Bioconjugate Chem., 2008, 19, 778–785 CrossRef CAS.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS.
- S. Melkko, Y. Zhang, C. E. Dumelin, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2007, 46, 4671–4674 CrossRef CAS.
- C. E. Dumelin, S. Trussel, F. Buller, E. Trachsel, F. Bootz, Y. Zhang, L. Mannocci, S. C. Beck, M. Drumea-Mirancea, M. W. Seeliger, C. Baltes, T. Muggler, F. Kranz, M. Rudin, S. Melkko, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2008, 47, 3196–3201 CrossRef CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS.
- J. L. Harris and N. Winssinger, Chem.–Eur. J., 2005, 11, 6792–6801 CrossRef CAS; N. Winssinger, J. L. Harris, B. J. Backes and P. G. Schultz, Angew. Chem., Int. Ed., 2001, 40, 3152–3154 CrossRef CAS; N. Winssinger, S. Ficarro, P. G. Schultz and J. L. Harris, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11139–11144 CrossRef CAS.
- J. Harris, D. E. Mason, J. Li, K. W. Burdick, B. J. Backes, T. Chen, A. Shipway, G. Van Heeke, L. Gough, A. Ghaemmaghami, F. Shakib, F. Debaene and N. Winssinger, Chem. Biol., 2004, 11, 1361–1372 CrossRef CAS; N. Winssinger, R. Damoiseaux, D. C. Tully, B. H. Geierstanger, K. Burdick and J. L. Harris, Chem. Biol., 2004, 11, 1351–1360 CrossRef CAS.
- F. Debaene, L. Mejias, J. L. Harris and N. Winssinger, Tetrahedron, 2004, 60, 8677–8690 CrossRef CAS; F. Debaene, J. Da Silva, Z. Pianowski, F. Duran and N. Winssinger, Tetrahedron, 2007, 63, 6577–6586 CrossRef CAS.
- H. D. Urbina, F. Debaene, B. Jost, C. Bole-Feysot, D. E. Mason, P. Kuzmic, J. L. Harris and N. Winssinger, ChemBioChem, 2006, 7, 1790–1797 CrossRef CAS.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Science, 1996, 274, 1531–1534 CrossRef CAS.
- I. Huc and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- O. Ramstrom and J. M. Lehn, Nature Reviews Drug Discovery, 2002, 1, 26–36 CrossRef CAS.
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells, Proc. Natl. Acad. Sci. USA, 2000, 97, 9367–9372 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Drug Discov. Today, 2002, 7, 117–125 CrossRef CAS.
- M. A. Koch, L. O. Wittenberg, S. Basu, D. A. Jeyaraj, E. Gourzoulidou, K. Reinecke, A. Odermatt and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16721–16726 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef.
- M. D. Burke, E. M. Berger and S. L. Schreiber, Science, 2003, 302, 613–618 CrossRef CAS.
- S. Pothukanuri, Z. Pianowski and N. Winssinger, Eur. J. Org. Chem., 2008, 3141–3148 CrossRef CAS.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Roper, K. B. Sharpless, C. H. Wong and H. C. Kolb, Angew. Chem., Int. Ed., 2005, 44, 116–120 CrossRef CAS.
- K. Gorska, K. T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS.
- K. Gorska, J. Beyrath, S. Fournel, G. Guichard and N. Winssinger, Chem. Commun., 2010, 46, 7742–7744 RSC.
- M. Beaulieu, G. P. Larson, L. Geller, S. D. Flanagan and T. G. Krontiris, Nucleic Acids Res., 2001, 29, 1114–1124 CrossRef CAS.
- While it is possible that some dissociation between the DNA template and PNA occurs under these conditions, the duplexes would reform upon cooling of the filtrates.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef.
- P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS.
- A. Miranker and M. Karplus, Proteins: Struct., Funct., Bioinf., 1991, 11, 29–34 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of PNA-encoded libraries, selection of library and resynthesized ligands. See DOI: 10.1039/c0sc00574f |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.