Selective anion binding by a “Chameleon” capsule with a dynamically reconfigurable exterior†
Yana R.
Hristova
,
Maarten M. J.
Smulders
,
Jack K.
Clegg
,
Boris
Breiner
and
Jonathan R.
Nitschke
*
University of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: jrn34@cam.ac.uk
First published on 23rd December 2010
Abstract
A new class of tetrahedral metal–organic capsules that can incorporate up to twelve different externally-directed amine residues is reported, allowing for very large dynamic libraries to be formed from mixtures of amines. Selectivity is observed both externally—more electron-rich amines are incorporated in favour of electron-poor amines—and internally—PF6− is bound in preference to CF3SO3− or BF4−.
Introduction
Container molecules1 are proving useful in a growing range of applications.2 One class of these molecules, tetrahedral metal–organic capsules,3–5 has been shown recently to catalyse reactions6 and to protect sensitive guests,7 releasing them to react upon receipt of a chemical signal. Mirroring the importance of sequestering reactive species in biological systems,8 the selective encapsulation and release of reagents within complex chemical systems9 will allow for the design of increasingly intricate ‘one-pot’ chemical transformations, as well as simple stimuli-responsive behaviour that may begin to mimic the much more complex adaptive behaviour of living systems.10New container molecules are needed to develop and explore these applications. Here we report the preparation and guest-binding studies of a new class of FeII4L6 tetrahedral hosts. These capsules were prepared via the subcomponent self-assembly approach,11,12 with twelve exchangeable aniline residues incorporated at the vertices of the tetrahedral cage. The modular construction of these cages allows for their exteriors to be tailored to suit a given environment and reconfigured in response to environmental stimuli; their well-defined central cavities allow selective guest binding.
Results and discussion
The reaction of 3,3′-bipyridine-6,6′-dicarboxaldehyde (6 equiv.) and aniline (12 equiv.) in the presence of an appropriate iron(II) salt (4 equiv.) led to the formation of the M4L6 cage complex 1 (Scheme 1). The 1H and 13C NMR spectra of capsule 1 display one set of ligand resonances in solution, consistent with T point symmetry. Acetonitrile solutions of 1 are deep purple, typical of the low-spin iron(II) tris(pyridylimine) chromophore.13 When 1 was prepared from Fe(BF4)2·6H2O or Fe(O3SCF3)2, the cage was observed to encapsulate one BF4− or CF3SO3− (OTf−) counterion by 19F NMR, which was confirmed by X-ray crystallographic analysis (Fig. 1).14 | ||
| Scheme 1 Subcomponent self-assembly of tetrahedral M4L6 cage complex 1; the structure of only one edge is shown for clarity. | ||
![Schematic representation of the crystal structure of [OTf− ⊂ 1]7+ (solvents and external anions removed for clarity).](/image/article/2011/SC/c0sc00495b/c0sc00495b-f1.gif) | ||
| Fig. 1 Schematic representation of the crystal structure of [OTf− ⊂ 1]7+ (solvents and external anions removed for clarity). | ||
There is an average distance of 9.5 Å between the metal centres. C2-symmetric bis-bidentate pyridylimine ligands define the edges of a tetrahedron, bridging the four six-coordinate FeII ions at each vertex. Each capsule is homochiral, with either ΛΛΛΛ or ΔΔΔΔ configurations at the metal centres and the OTf− guest is completely encapsulated within the cage.
When p-chloroaniline was used in place of unsubstituted aniline in the reaction of Scheme 1, the analogous capsule 2 was formed (Scheme 2) containing 12 p-chloroaniline residues. We have previously reported the substitution of electron-poor aniline residues by more electron-rich anilines within copper(I) complexes.12,15 To explore this phenomenon in the present system, the reactions of 2 with p-toluidine (14.7 equiv.) and p-methoxyaniline (12.5 equiv.) were investigated (Scheme 2). The addition of either of these two more electron-rich anilines to 2 resulted in the quantitative displacement of the p-chloroaniline residues of 2 and the formation of p-toluidine-containing 3 and p-methoxyaniline-containing 4, respectively (Scheme 2), as observed by 1H NMR (Figures S3, S4†).
 | ||
| Scheme 2 Subcomponent substitution driven by electronic effects: more electron-rich anilines are able to displace more electron-poor anilines. | ||
When p-methoxyaniline was added to a solution of 3, its transformation into 4 was incomplete, resulting in only 79% substitution (Scheme 2). ESI-MS analysis revealed the presence of a mixture of products with different degrees of substitution ranging from only two p-methoxyanilines to all twelve p-methoxyanilines being incorporated (Figures S5, S6†). We attribute this incomplete substitution to the smaller difference in electronic properties between p-Me and p-OMe substituents, as compared to the larger difference between either of these two electron-rich substituents and electron-withdrawing p-Cl.
The iron(II)-templated reaction of 3,3′-bipyridine-6,6′-dicarboxaldehyde (6 equiv.) with p-bromoaniline, p-chloroaniline and p-iodoaniline (∼4 equiv. each) resulted in the formation of a far larger library of cages (Scheme 3). Six potential ligands (three homotopic and three heterotopic) could be formed and these ligands could be combined to give 91 possible cage products different by mass and hundreds of thousands more if the many possible structural isomers with identical masses were taken into account. It was possible to record a 1H NMR spectrum of this mixture (Fig. 2a) as it retained the diamagnetism exhibited by 1. ESI-MS could be employed to further characterise the system (Figure S2†). A multitude of +3 and +4 charged cage species with masses that could be assigned to multiply substituted species (due to the mass differences of the halogens present) was observed.
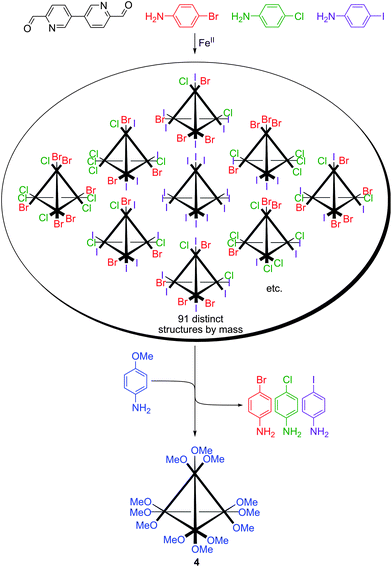 | ||
| Scheme 3 A library of heteroleptic tetrahedral cages and its transformation to a single homoleptic cage 4 upon aniline substitution. | ||
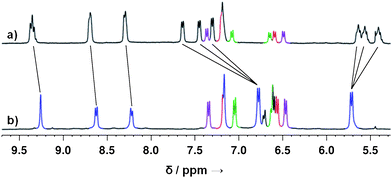 | ||
| Fig. 2 a) The aromatic region of the 1H NMR spectrum of the library of heteroleptic (coloured peaks represent excess anilines); b) 1H NMR spectrum after subcomponent substitution by p-methoxyaniline (blue signals correspond to homoleptic cage 4). | ||
The addition of twelve equivalents of the more electron-rich p-methoxyaniline to the mixture depicted in Scheme 3 resulted in the collapse of the library into the single homoleptic structure 4. This transformation is clearly observed by 1H NMR (Fig. 2): the multiple peaks at 9.35 ppm (imine protons) were observed to transform into a sharp imine singlet at 9.26 ppm, the three doublets between 7.20 and 7.70 ppm became a single doublet at 6.78 ppm, and the three sets of multiple peaks between 5.37–5.64 ppm became a sharp doublet at 5.71 ppm. In agreement with the 1H NMR spectrum, 4 was the only product observed by ESI-MS.
As discussed above, 1 was found to be a suitable host for a triflate guest. The cages of the library of Scheme 3 were also observed to encapsulate triflate in solution (by 19F NMR) and in the ESI mass spectrum each cage was associated with an OTf− ion (Figure S2†). Both BF4− and PF6− were also found to be encapsulated by 1–4 by 19F NMR. The binding was found to be selective; when an excess of tetramethylammonium hexafluorophosphate was added to [BF4− ⊂ 1] the encapsulated BF4− was displaced by the larger PF6− anion (Figure S7†). Analogously, when [OTf− ⊂ 2] was treated with excess potassium hexafluorophosphate, a displacement of OTf− by PF6− was again observed (Figure S8†). Peripheral substitution patterns thus do not appear qualitatively to impact guest binding preferences, with capsules containing both electron-rich and electron-poor aniline residues following the anion binding trend of BF4− < OTf− < PF6−.
Substantial recent attention has been paid to selective anion-binding species.16 Quantification of anion-binding strength17 is required in order to gauge a host's utility and selectivity. Several M4L6 tetrahedra have been demonstrated to selectively encapsulate anions within their cavities.4,5,18 However, determining binding constants has proven difficult; some cages are difficult to isolate in the absence of a template guest, necessitating the application of novel techniques19 to determine guest binding strength. We undertook, thus, to synthesise cage 1 with a larger counteranion with the aim of obtaining an anion-free cavity for binding studies.5 Cage 1 has a calculated guest-accessible void volume of 136 Å3.20 The trifluoromethanesulfonimide (triflimide, NTf2−) counteranion was thus chosen, as its volume (157 Å3) would render it too large to serve as a guest for 1. The use of Fe(NTf2)2 in the procedure of Scheme 1 enabled the isolation of cage 1 containing no anion, as confirmed by single crystal X-ray diffraction (Fig. 3).11 A single acetonitrile solvent molecule was found within the cavity, which was not observed by NMR in solution. This result confirmed that an anionic template is not required for cage formation.4
![Schematic representation of the crystal structure of [MeCN ⊂ 1]8+ (hydrogens, solvents and anions removed for clarity).](/image/article/2011/SC/c0sc00495b/c0sc00495b-f3.gif) | ||
| Fig. 3 Schematic representation of the crystal structure of [MeCN ⊂ 1]8+ (hydrogens, solvents and anions removed for clarity). | ||
The successful isolation of “empty” 1 (i.e.1 without an anionic guest) enabled the quantitative determination of the anion-binding constants shown in Table 1. As noted above, BF4−, OTf−, and PF6− all proved to be suitable guests for 1, however, no evidence was observed for the encapsulation of small neutral molecules such as methane, ethane, carbon dioxide, acetonitrile, dichloromethane or sulfur hexafluoride in solution.
| Complex | NMR Exchange | V (Å3) | % of hostV | K a (M−1) |
|---|---|---|---|---|
| [BF4− ⊂ 1]7+ | Fast | 59 | 43% | 2.3(4) × 104 |
| [OTf− ⊂ 1]7+ | Slow | 86 | 63% | 5.2(8) × 104 |
| [PF6− ⊂ 1]7+ | Slow | 75 | 55% | 1.3(3) × 106 |
The strength of the observed anion binding reflects the deviation of the anions' volumes from Rebek's optimum of 55% cavity occupation21 (see Figure S9 for size comparisons; Ka determination procedures are given in the ESI†). The affinity of 1 for PF6− was exceptionally high, representing the strongest host–guest binding of which we are aware for a metal–organic host that is isolable in the absence of a guest.19,22 Because of its high binding strength, the Ka value of PF6− could not be derived with precision directly by NMR titration, requiring a competitive binding experiment based on the displacement of OTf− by PF6−. The Ka values obtained confirm the qualitative observations that PF6− quantitatively displaced either BF4− or OTf− and that BF4− and OTf− coexist as guests in equilibrium with each other; the latter mirrors the substitution patterns of anilines with different affinities for incorporation.
Conclusions
The two levels of selective dynamic interchange demonstrated by this system—at the periphery, through aniline exchange, and at the centre, through guest exchange, are mutually orthogonal. Such capsules might thus be fitted with externally-directed groups permitting solubility in a given solvent system, or possibly which facilitate the recognition and binding to a given target in a biological system. The guest within could then diffuse out, either in response to a specific signal such as a competitive guest, or simply as part of its equilibration process. Larger versions of the system reported here could thus serve as subsystems within larger systems that are capable of providing complex responses to applied stimuli.23We thank Dr J. E. Davies for collecting the X-ray data, Dr G. D. Pantoş for helpful discussions, D. Lambin and C. Sporikou for the synthesis of 3,3′-bipyridine-6,6′-dicarboxaldehyde. Mass spectra were provided by the EPSRC National MS Service Centre at Swansea. This work was supported by the Cambridge European Trust (Y. R. H.), the Netherlands Organisation for Scientific Research-NWO (M. M. J. S.), the Marie Curie IIF scheme of the 7th EU Framework Program (J. K. C.), the Isaac Newton Trust (B. B.), the Walters-Kundert Charitable Trust (J. R. N.) and the EPSRC.
References
- D. J. Cram, Nature, 1992, 356, 29–36 CrossRef CAS; S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS; F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Jr., Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; E. Botana, E. Da Silva, J. Benet-Buchholz, P. Ballester and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 198–201 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS; B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC; I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC; L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2004, 126, 14366–14367 CrossRef CAS.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS; J. K. Clegg, L. F. Lindoy, B. Moubaraki, K. S. Murray and J. C. McMurtrie, Dalton Trans., 2004, 2417–2423 RSC; M. Albrecht and R. Froehlich, Bull. Chem. Soc. Jpn., 2007, 80, 797–808 CrossRef CAS; R. W. Saalfrank, R. Burak, A. Breit, D. Stalke, R. Herbst-Irmer, J. Daub, M. Porsch, E. Bill, M. Muether and A. X. Trautwein, Angew. Chem., 1994, 106, 1697–1699 CrossRef CAS; D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS.
- M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC.
- C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 1190–1192 RSC.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS; M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371–380.
- R. Warmuth, Angew. Chem. Int. Ed., 1997, 36, 1347–1350 CrossRef CAS; T. Iwasawa, R. J. Hooley and J. Rebek, Jr., Science, 2007, 317, 493–496 CrossRef CAS; P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS; K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS.
- A. Dautry-Varsat, A. Ciechanover and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 2258–2262 CAS; E. C. Theil, Annu. Rev. Biochem., 1987, 56, 289–315 CrossRef CAS.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC; J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS; G. Ashkenasy, R. Jagasia, M. Yadav and M. R. Ghadiri, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10872–10877 CrossRef CAS; A. Vidonne and D. Philp, Eur. J. Org. Chem., 2009, 593–610 CrossRef CAS; L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287–302 RSC; L. Zelikovich, J. Libman and A. Shanzer, Nature, 1995, 374, 790–792 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, 2000 Search PubMed; J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- V. E. Campbell and J. R. Nitschke, Synlett, 2008, 3077–3090 CAS; M. Hutin, G. Bernardinelli and J. R. Nitschke, Chem.–Eur. J., 2008, 14, 4585–4593 CrossRef CAS.
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS.
- J.-H. Wei and C.-S. Hsiao, J. Inorg. Nucl. Chem., 1981, 43, 2299–2300 CrossRef.
- Details of the structural characterisation are given in the supporting information.
- D. Schultz and J. R. Nitschke, J. Am. Chem. Soc., 2006, 128, 9887–9892 CrossRef CAS.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, 2006 Search PubMed; S. R. Bayly and P. D. Beer, Struct. Bonding, 2008, 129, 45–94 Search PubMed; S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 CAS.
- M. J. McDonough, A. J. Reynolds, W. Y. G. Lee and K. A. Jolliffe, Chem. Commun., 2006, 2971–2973 RSC; M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366–375 RSC.
- R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025–4029 CrossRef CAS.
- G. J. Kleywegt and T. A. Jones, Acta. Cryst., 1994, D50, 178–185 CAS.
- S. Mecozzi and J. Rebek, Jr., Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- C. Sgarlata, J. S. Mugridge, M. D. Pluth, B. E. F. Tiedemann, V. Zito, G. Arena and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 1005–1009 CrossRef CAS.
- V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684–687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis, NMR spectra, X-ray measurements, binding constant measurements and calculations; interview with Jonathan R. Nitschke. CCDC reference numbers 782794 and 787146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00495b |
| This journal is © The Royal Society of Chemistry 2011 |