Dehydration triggered asymmetric hydrogenation of 3-(α-hydroxyalkyl)indoles†
Duo-Sheng
Wang
a,
Jie
Tang
b,
Yong-Gui
Zhou
*a,
Mu-Wang
Chen
a,
Chang-Bin
Yu
a,
Ying
Duan
a and
Guo-Fang
Jiang
*b
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, P. R. China. E-mail: ygzhou@dicp.ac.cn
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: guofangjiang@yahoo.com.cn
First published on 24th January 2011
Abstract
Highly enantioselective hydrogenation of 3-(α-hydroxyalkyl)indoles promoted by a Brønsted acid for dehydration to form a vinylogous iminium intermediate in situ was developed with Pd(OCOCF3)2/(R)-H8-BINAP as catalyst with up to 97% ee. This methodology provides an efficient and rapid access to chiral 2,3-disubstituted indolines.
Catalytic asymmetric hydrogenation of prochiral compounds, such as olefins, ketones and imines, is one of the well-established reactions and holds a venerable position in organic synthesis.1 Nevertheless, the asymmetric hydrogenation of aromatic and heteroaromatic compounds had been unexplored until recently.2 This may mainly ascribe to the high stability of these compounds and harsh conditions needed to destroy the aromaticity which adversely affects the enantioselectivity.3 Consequently, activation strategies for both catalyst and substrate emerged as the solution for the successful asymmetric hydrogenation of aromatic compounds. Based on this principle, some transition-metal and organic catalysts have been successfully introduced to the asymmetric hydrogenation2 and transfer hydrogenation4 of heteroaromatic compounds. Heretofore, some breakthroughs have been achieved in the asymmetric reduction of heteroaromatic compounds such as quinolines, quinoxalines, pyridines, indoles, pyrroles and furans.5,6 Despite advances, it was far from meeting the continuously expansive requirement of the corresponding chiral saturated compounds in pharmaceutical and agrochemical synthesis.7 Some heteroaromatic compounds even have been eluded for asymmetric hydrogenation.
In 2003, our group reported the first asymmetric hydrogenation of quinolines employing iodine as an additive to activate the iridium catalysts.8a Thereafter, this strategy was extensively employed in the hydrogenation of quinolines as well as pyridines8c and quinoxalines by us and others. Then, chloroformate was applied as substrate activator for asymmetric hydrogenation of quinolines as well as isoquinolines by us.8b Very recently, we described the asymmetric hydrogenation of quinolines and simple indoles with catalytic and stoichiometric amount of Brønsted acid as activator, respectively.8d,9 In our ongoing efforts toward the development of asymmetric hydrogenation of aromatic compounds, we became interested in exploring new substrate activation strategies.
Easily available 3-(α-hydroxyalkyl)indoles can readily dehydrate to form vinylogous iminium intermediates in situ in the presence of Brønsted acids, which have been successfully applied to some chemical transformations.10 We envision that the active vinylogous iminium intermediate should be easily hydrogenated with a proper catalytic system since the aromaticity has been partially destroyed. This partial dearomatization process triggered by dehydration offered a new opportunity to the asymmetric hydrogenation of 3-(α-hydroxyalkyl)indoles (Scheme 1). In this communication, asymmetric hydrogenation of 3-(α-hydroxyalkyl)indoles is successfully developed with up to 97% ee.
 | ||
| Scheme 1 Dehydration triggered partial dearomatization of 3-(α-hydroxyalkyl)indoles for hydrogenation. | ||
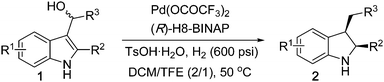
A series of racemic 2,3-disubstituted 3-(α-hydroxyalkyl)indoles was synthesized rapidly and conveniently through a divergent approach starting from the formylation of 2-substituted indoles followed by nucleophilic additions with various Grignard Reagents.10h,11
Obviously, both acid and water were existing in this system, and accordingly the asymmetric hydrogenation catalysts must be compatible with both acid and water. Recently, chiral palladium catalysts have been successfully applied to asymmetric hydrogenation of activated imines, simple indoles and functionalized ketones by us and other groups.9,12 Our preliminary mechanistic study found the chiral palladium catalyst is not sensitive to acid and water. Therefore, chiral palladium catalysts should be a good choice for the asymmetric hydrogenation of 3-(α-hydroxyalkyl)indoles.
Initially, (2-methyl-1H-indol-3-yl)(phenyl)methanol 1a was selected as a model substrate to test our hypothesis. Pd(OCOCF3)2/(R)-BINAP was employed as catalyst and L-camphorsulfonic acid (L-CSA) as the activator, the hydrogenation reaction was conducted in a mixture of solvents DCM/TFE (dichloromethane and 2,2,2-trifluoroethanol: 1/1) at 50 °C.9 The reaction proceeded smoothly to give the desired product 2a with 86% yield and 86% ee (Table 1, entry 1). Catalytic amount of activator was tested but with low yield and decreased ee (entry 2). Mixed solvents of DCM and TFE with different ratios were examined (entries 3–6), and the best result was obtained with the ratio of 2/1 in terms of enantioselectivity (entry 3, 88% ee). Next, the effect of different acid activators on the reactivity and enantioselectivity was examined (entries 7–10). It was found that strong Brønsted acid was necessary for obtaining the desired product 2a (entries 7–9). When weak acid, benzoic acid, was applied in this transformation, no desirable product was obtained (entry 10). Commercially available p-toluenesulfonic acid monohydrate (TsOH·H2O) gave both high yield and enantioselectivity, and it was selected for further studies (entry 7, 87% ee). Ligand screen indicated that (R)-H8-BINAP gave the highest enantioselectivity (entry 11, 91% ee). Therefore, the optimal conditions were established as the following: Pd(OCOCF3)2/(R)-H8-BINAP/TsOH·H2O/H2 (600 psi)/DCM-TFE (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/50 °C.
:1)/50 °C.
a

| Entry | Solvent | Acid | Yield (%) | ee (%)b |
|---|---|---|---|---|
| a Conditions: 0.25 mmol 1a, Pd(OCOCF3)2 (2 mol%), (R)-BINAP (2.4 mol%), acid (0.25 mmol), 3 mL solvent, 50 °C, 16–24 h. b Determined by HPLC. c With 0.05 mmol L-CSA (0.2 equiv). d With full conversion of 1a and 2-methyl-3-benzylindole 3 was obtained as byproduct. e With (R)-H8-BINAP as ligand. | ||||
| 1 | DCM/TFE (1/1) | L-CSA | 86 | 86 |
| 2c | DCM/TFE (1/1) | L-CSA | 20d | 71 |
| 3 | DCM/TFE (2/1) | L-CSA | 75 | 88 |
| 4 | DCM | L-CSA | 58d | 57 |
| 5 | DCM/TFE (1/2) | L-CSA | 79 | 79 |
| 6 | TFE | L-CSA | 68 | 86 |
| 7 | DCM/TFE (2/1) | TsOH·H2O | 91 | 87 |
| 8 | DCM/TFE (2/1) | TfOH | 97 | 77 |
| 9 | DCM/TFE (2/1) | TFA | 81 | 43 |
| 10 | DCM/TFE (2/1) | PhCO2H | —d | — |
| 11e | DCM/TFE (2/1) | TsOH·H2O | 96 | 91 |
| 12 | DCM/TFE (2/1) | D-CSA | 74 | 78 |
Various 3-(α-hydroxyalkyl)indoles were subjected to the Pd-catalyzed asymmetric hydrogenation as shown in Table 2. For 2-methyl substituted substrates 1a–1h, when R3 is an aryl group, excellent enantioselectivities (88–91% ee) were obtained regardless of position and electronic effect of substituents of phenyl ring (entries 1–6). When R3 is an alkyl group, slightly higher enantioselectivities were obtained (entries 7–8). For 2-butyl and 2-phenethyl substituted indoles 1i–1l, 93–96% ee were obtained (entries 9–12). Substitution at the 5-position gave negative effect on enantioselectivity which may ascribe to both steric and electronic effect (entries 13–14). Excellent 94–97% ee were achieved with substrates assembled with a methyl substituent at 7-position (entries 15–20), and this may ascribe to the steric effect of 7-methyl.
 | (1) |
 | (2) |
a
| Entry | R1 | R2 | R3 | Yield (%) | ee (%)b |
|---|---|---|---|---|---|
| a Conditions: 0.25 mmol 1, Pd(OCOCF3)2 (2 mol%), (R)-H8-BINAP (2.4 mol%), TsOH·H2O (0.25 mmol), 3 mL solvent, 50 °C, 16–24 h. b Determined by HPLC. | |||||
| 1 | H | Me | Ph | 96 (2a) | 91 |
| 2 | H | Me | 4-MeC6H4 | 97 (2b) | 90 |
| 3 | H | Me | 3-MeC6H4 | 86 (2c) | 90 |
| 4 | H | Me | 2-MeC6H4 | 85 (2d) | 91 |
| 5 | H | Me | 4-MeOC6H4 | 86 (2e) | 89 |
| 6 | H | Me | 4-FC6H4 | 88 (2f) | 88 |
| 7 | H | Me | Cy | 95 (2g) | 94 |
| 8 | H | Me | i-Pr | 99 (2h) | 94 |
| 9 | H | n-Bu | Ph | 94 (2i) | 94 |
| 10 | H | n-Bu | Cy | 96 (2j) | 96 |
| 11 | H | Phenethyl | Ph | 85 (2k) | 93 |
| 12 | H | Phenethyl | Cy | 78 (2l) | 95 |
| 13 | 5-F | Me | Ph | 92 (2m) | 85 |
| 14 | 5-F | Me | Cy | 91 (2n) | 88 |
| 15 | 7-Me | Me | Ph | 84 (2o) | 97 |
| 16 | 7-Me | Me | 4-MeC6H4 | 94 (2p) | 96 |
| 17 | 7-Me | Me | 3-MeC6H4 | 92 (2q) | 95 |
| 18 | 7-Me | Me | 2-MeC6H4 | 99 (2r) | 94 |
| 19 | 7-Me | Me | Cy | 98 (2s) | 97 |
| 20 | 7-Me | Me | i-Pr | 98 (2t) | 97 |
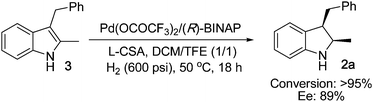
The hydrogenation reaction was driven by Brønsted acid promoting dehydration to form a vinylogous iminium in situ. To obtain information on the hydrogenation reaction of vinylogous iminium intermediate, we performed hydrogenation reaction of 3-(α-hydroxyalkyl)indole 1a at room temperature using the chiral palladium complex as catalyst (eqn (1)). The desirable product 2a was obtained with 91% ee, and indole 3 was the main product (2a/3 = 26/74). When 3 was subjected to hydrogenation at 50 °C (eqn (2)), full conversion and 89% ee were obtained. These results indicated that indole 3 might be the intermediate for the 3-(α-hydroxyalkyl)indole hydrogenation.9
By analysis of Pd-catalyzed hydrogenation sequence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds of vinylogous iminium intermediate, we envisioned that there might be three possible paths.13 Path A: 1,2-hydride addition and isomerization to form indole, and then acid activated indole hydrogenation. Path B: 3,4-hydride addition and isomerization to form indole, as well as acid activated indole hydrogenation. Path C: 1,4-hydride addition to form indole followed by acid activated indole hydrogenation. To shed light on the reaction mechanism, we performed three isotopic labeling experiments with deuterated solvent and deuterium gas, respectively (Scheme 2).13 When the hydrogenation was carried out in deuterated TFE for 18.5 h, full conversion was attained with 2a as the product. One deuterium atom was detected at the 3-position with 94% incorporation (eqn 3). When the hydrogenation reaction was stopped at 2.5 h, 3 and 2a were obtained at a ratio of 85/15; 1H NMR analysis of the isolated indole 3 showed no deuterium atom was incorporated at the benzylic position (eqn 4). When 1a was treated with D2, 2a was obtained with 97% and 91% incorporation of deuterium at 2- and benzylic position, respectively (eqn 5).
C and CN bonds of vinylogous iminium intermediate, we envisioned that there might be three possible paths.13 Path A: 1,2-hydride addition and isomerization to form indole, and then acid activated indole hydrogenation. Path B: 3,4-hydride addition and isomerization to form indole, as well as acid activated indole hydrogenation. Path C: 1,4-hydride addition to form indole followed by acid activated indole hydrogenation. To shed light on the reaction mechanism, we performed three isotopic labeling experiments with deuterated solvent and deuterium gas, respectively (Scheme 2).13 When the hydrogenation was carried out in deuterated TFE for 18.5 h, full conversion was attained with 2a as the product. One deuterium atom was detected at the 3-position with 94% incorporation (eqn 3). When the hydrogenation reaction was stopped at 2.5 h, 3 and 2a were obtained at a ratio of 85/15; 1H NMR analysis of the isolated indole 3 showed no deuterium atom was incorporated at the benzylic position (eqn 4). When 1a was treated with D2, 2a was obtained with 97% and 91% incorporation of deuterium at 2- and benzylic position, respectively (eqn 5).
 | ||
| Scheme 2 Deuterium-labeling studies. | ||
With path A, when deuterium gas is used, only one deuterium atom should be incorporated to the 2 position of indoline 2. The incorporation of two deuterium atoms in the product excludes this path (Scheme 2, eqn 5). For the paths B and C, isotopic labeling experiments cannot completely differentiate. Path C is more favorable in thermodynamics than path B because of rapid recovery of aromatization. Hence, the hydrogenation sequence of the CC and CN bonds of the iminium intermediate could be determined as 1,4-hydride addition followed by 1,2-hydride addition (Scheme 3). The first hydrogen molecule could be readily added to the vinylogous iminium through 1,4-hydride addition to form 2,3-disubstituted indole and thus recovered its aromaticity. The further hydrogenation of the indole must be activated by strong Brønsted acid to form an iminium intermediate, which was hydrogenated via1,2-hydride addition as disclosed in our previous work.9
 | ||
| Scheme 3 Proposed process of 3-(α-hydroxyalkyl)indoles hydrogenation. | ||
In summary, we have developed an efficient activation strategy for the Pd-catalyzed asymmetric hydrogenation of 3-(α-hydroxyalkyl)indoles with a Brønsted acid as an activator. Dehydration is the driving force for the reductive removal of the hydroxy group with the formation of vinylogous iminium intermediate. The starting racemic 3-(α-hydroxyalkyl)indoles are readily accessible and this methodology provides an efficient and rapid access to the chiral 2,3-disubstituted indolines.
Acknowledgements
We are grateful to Financial support from National Science Foundation of China (20921092 & 21032003), National Basic Research Program of China (2010CB833300) and Dalian Institute of Chemical Physics (K2010F1).Notes and references
-
(a)
P. N. Rylander, Catalytic Hydrogenation in Organic Synthesis, Academic Press, New York, 1979, p. 175 Search PubMed
; (b) E. N. Jacobsen, A. Pfaltz, H. Yamamoto; ed. Comprehensive Asymmetric Catalysis; Springer: Berlin, 1999 Search PubMed
- For recent reviews on asymmetric hydrogenation of aromatic and heteroaromatic compounds, see:
(a) P. J. Dyson, Dalton Trans., 2003, 2964 RSC
- C. W. Bird, Tetrahedron, 1992, 48, 335 CrossRef CAS
- For recent reviews on transfer hydrogenation mediated by Hantzsch esters, see:
(a) S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS
- Selected examples on asymmetric reduction of heteroaromatics, quinolines:
(a) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef
- For asymmetric hydrogenation of indoles, see:
(a) R. Kuwano, K. Sato, T. Kurokawa, D. Karube and Y. Ito, J. Am. Chem. Soc., 2000, 122, 7614 CrossRef CAS
-
D. Barton, K. Nakanishi, O. Meth-Cohn, Comprehensive Natural Products Chemistry, Elsevier, Oxford, 1999, Vol. 1–9 Search PubMed
- Selected examples of our group's work on asymmetric hydrogenation of heteroaromatics, see:
(a) W.-B. Wang, S.-M. Lu, P.-Y. Yang, X.-W. Han and Y.-G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536 CrossRef CAS
- D.-S. Wang, Q.-A. Chen, W. Li, C.-B. Yu, Y.-G. Zhou and X. Zhang, J. Am. Chem. Soc., 2010, 132, 8909 CrossRef CAS
- Selected examples on transformations involved vinylogous iminium intermediates, see:
(a) N. A. Kogan and G. N. Kulbitskii, Chem. Heterocycl. Compd., 1978, 14, 46 CrossRef
-
(a) M. Mor, S. Rivara, C. Silva, F. Bordi and P. V. Plazzi, J. Med. Chem., 1998, 41, 3831 CrossRef CAS
- Selected examples on Pd catalyzed asymmetric hydrogenation, our group's work, see:
(a) Y.-Q. Wang, S.-M. Lu and Y.-G. Zhou, Org. Lett., 2005, 7, 3235 CrossRef CAS
- For detailed information about the study of mechanism, see the Supporting Information.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization of new compounds. See DOI: 10.1039/c0sc00614a |
| This journal is © The Royal Society of Chemistry 2011 |