Nanostructures of tetranuclear copper(I) complexes with short Cu(I)⋯Cu(I) contacts: crystallization-induced emission enhancement†
Yong
Chen
ab,
Jun-Li
Li
c,
Glenna So Ming
Tong
b,
Wei
Lu
b,
Wen-Fu
Fu
*ac,
Siu-Wai
Lai
b and
Chi-Ming
Che
*b
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: fuwf@mail.ipc.ac.cn
bDepartment of Chemistry, Institute of Molecular Functional Materials, and HKU-CAS Joint Laboratory on New Materials, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: cmche@hku.hk
cCollege of Chemistry and Engineering, Yunnan Normal University, Kunming, 650092, P. R. China
First published on 10th June 2011
Abstract
Tetranuclear copper(I) complexes containing N,N′-bis(5,7-dimethyl-1,8-naphthyridine-2-yl)amine and phosphine ligands with close intramolecular Cu(I)⋯Cu(I) contacts were found to precipitate as thermodynamically stable and crystalline quasi-2D sheet-like nanostructures. Kinetically stable, amorphous, spherical particles were also identified during the precipitation/crystallization processes of these copper(I) complexes in a dichloromethane/hexane mixture. The distinct phosphorescent properties of these two forms of nanostructures were studied and a crystallization-induced emission enhancement was observed during the morphological evolution from amorphous spherical particles to crystalline nanosheets. All of these photophysical properties were rationalized by density functional theory calculations.
Introduction
Electronic and catalytic properties of inorganic nanostructures are highly dependent on their size and morphology.1 For organic nanostructures, as their excitons are usually confined to a few nanometres, their photophysical properties generally display a dependence on intermolecular interactions.2 Despite the remarkable progress in inorganic and purely organic nanomaterials, nanostructures with well-defined morphologies made from transition metal complexes are sparse. Our interest in nanostructures that include both metal ions and organic ligands is focused on the development of functional materials that combine the features of purely inorganic and organic nanomaterials. Nanostructures of low-molecular-weight metal complexes can be constructed through weak intermolecular hydrogen bonding, π–π stacking, metal⋯metal interactions and van der Waals forces.3–5 For example, quasi-one-dimensional (quasi-1D) nanostructures of tris-(8-hydroxyquinoline) aluminum (Alq3) have been prepared by physical vapor deposition methods or by surfactant-assisted solution routes and their optical and electronic properties have been evaluated.3a,b Shelnutt and co-workers fabricated metalloporphyrin nanostructures with a well-defined shape and size through both ionic self-assembly and reprecipitation methods.4 We and others have also obtained functionalized nanomaterials from square-planar platinum(II)5a–i and rhodium(I)5j,k complexes, wherein intermolecular metal⋯metal interactions play a key role in directing the anisotropic growth of the nanostructures.Luminescent polynuclear copper(I) complexes have been extensively studied for their diverse structures and rich photophysical and photochemical properties.6 In recent years, there have been a few reports on the application of luminescent copper(I) complexes in organic light emitting diodes.7 However, supramolecular nanostructures from low-molecular-weight copper(I) complexes are surprisingly yet to be explored. We are particularly interested in the photophysical properties and self-assemblies of copper(I) complexes with intramolecular Cu(I)⋯Cu(I) contacts.8 In the present work, two tetranuclear copper(I) complexes (1 and 2, Scheme 1) with short Cu(I)⋯Cu(I) contacts were found to display rich photophysical properties and fluxional behaviors. Quasi-two-dimensional (quasi-2D) sheet-like nanostructures could be fabricated from these copper(I) complexes by a simple reprecipitation method and spherical particles have been identified as an intermediate during the precipitation/crystallization processes of these copper(I) complexes in a dichloromethane/hexane mixture. We found that the emission from these nanostructures is morphology-dependent. A crystallization-induced emission enhancement (CIEE)9 was observed in the present copper(I) system.
 | ||
| Scheme 1 The synthesis and chemical structures of 1–3. | ||
Results and discussion
Synthesis and characterization
The copper(I) complexes 1–3 were prepared by the reactions of [Cu(CH3CN)4](BF4), N,N′-bis(5,7-dimethyl-1,8-naphthyridine-2-yl)amine (HL)10 and the corresponding phosphine ligands (1,1-bis(dicyclohexylphosphino)methane (dcpm) for 1; 1,1-bis(diphenylphosphino)methane (dppm) for 2; and PPh3 for 3) under a nitrogen atmosphere. Mononuclear and tetranuclear copper(I) complexes were obtained when monodentate and bidentate phosphine ligands were used, respectively. All of the complexes were characterized by elemental analyses, FAB-MS (fast-atom-bombardment mass spectrometry) and various spectroscopic techniques. Their solid state structures were determined by single crystal X-ray analysis.‡Thermal gravimetric analysis (TGA) revealed that powder samples of 1 and 2 are thermally stable up to 300 °C under a nitrogen atmosphere (see ESI†).Complex 1 crystallized in a monoclinic space group. Four copper atoms are coordinated by two naphthyridine ligands and two diphosphine ligands (Figs 1a and b). Each Cu atom has a trigonal planar geometry, with N–Cu–P angles from 116.13(4) to 133.61(4)° and a mean N–Cu–N angle of 110.22(6)°. The outer Cu(2) and Cu(2A) are coordinated by two nitrogen atoms from two naphthyridine rings, with Cu–N distances of 2.0608(1) and 2.0338(1) Å, whereas the inner Cu(1) and Cu(1A) are bonded to a basic amido N atom together with a naphthyridyl N atom. Notably, the short distance of 2.688 Å between Cu(1) and N(1A) reveals a weak Cu–N interaction. The Cu(1)⋯Cu(2) and Cu(1)⋯Cu(1A) distances of 2.6116 and 3.103 Å, respectively, are shorter than or close to the sum of the van der Waals radii of two copper atoms (2.8 Å).11
 | ||
| Fig. 1 Crystal structures for the cations of (a and b) 1 and (d and e) 2 showing 30% probability ellipsoids with the hydrogen atoms omitted for clarity. Variable-temperature 1H and 31P NMR spectra of (c) 1 and (f) 2 in CD2Cl2 solutions. | ||
Single crystal X-ray analysis revealed that the complex cations in 1 and 2 (Figs 1d and e) have identical coordination modes at the copper(I) ions, except for the intramolecular Cu⋯Cu contacts. The dppm ligand is less bulky than dcpm, resulting in shorter intramolecular Cu⋯Cu distances of 2.6107 and 2.8223 Å and larger N–Cu–N angles of 118.15 and 118.58°. For the mononuclear complex 3, the Cu atom is coordinated by a phosphine atom from PPh3 and two nitrogen atoms from naphthyridyl rings, where HL acts as a bidentate ligand (see ESI†).
As revealed from the single crystal structure data of 1 and 2, the two naphthyridine rings in ligand HL have different coordination modes, and hence should not be chemically equivalent. However, the ambient temperature NMR spectra of complexes 1 (Fig. 1c) and 2 (Fig. 1f) display only three well-resolved signals for naphthyridine ring protons in the aromatic region and two single peaks for methyl protons, revealing that all four naphthyridine rings are equivalent on the NMR time scale. Fig. 1c displays the 1H NMR spectra of complex 1 at 20, 0, −20 and −40 °C. When the temperature decreases from 20 to −40 °C, three resonances in the aromatic region first broaden (at ca. −20 °C) and then split (at ca. −40 °C) into six signals. The peaks of the methyl protons show similar changes, while the 31P NMR spectra of 1 at these temperatures are the same. These observations reveal that complex 1 retains the same structure in a low-temperature solution as in the solid state. While at elevated temperature, the naphthyridine rings turn out to be chemically equivalent by a fluxional process, in which the ligand HL rapidly exchanges its points of attachment to Cu atoms (see ESI†). We note that such a coordination site exchange has been observed for several other mononuclear 1,8-naphthyridine complexes.12
Supramolecular nanostructures
Superstructures of complexes 1–3 were obtained by a reprecipitation method without the aid of surfactants. Typically, the complex was dissolved in dichloromethane to give a bright or pale yellow solution (ca. 1.5 mM, 0.1 mL), and then n-hexane (3 mL) was added to the dichloromethane solution, resulting in a yellow suspension. After aging for 24 h, the suspensions of complexes 1–3 were transferred to a silica wafer or a carbon-coated copper grid, and examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM and TEM images revealed that the suspensions of complexes 1 and 2 contained sheet-like nanostructures (Fig. 2). Generally, both the length and width of these quasi-2D structures ranged from hundreds of nanometres to several microns, with a thickness of around 70 nanometres (the cross-section was observed under SEM, see ESI†). In contrast to those of complexes 1 and 2, suspensions of complex 3 consisted of microrods (see ESI†). The selected area electron diffraction (SAED) pattern of a single nanosheet and a microrod showed sharp and ordered spots, revealing that these nanostructures are crystalline in nature. For complex 1, the lateral d-spacings of 12.6 and 13.1 Å observed in the SAED pattern could be indexed according to the X-ray single crystal structure as the respective [1 1–1] and [200] Miller planes. | ||
| Fig. 2 SEM (a and d), TEM (b and e) and the corresponding SAED (c and f) images of nanostructures 1 (a, b and c) and 2 (d, e and f). | ||
To track the growth process of these crystalline superstructures in the mixed solvent system, time course experiments were conducted and examined by electron microscopy. Complex 1 is taken as an example in the following description. After a reaction time of one hour, only spherical particles were observed (Fig. 3a), the sizes of which varied from tens of nanometres to several hundred nanometres. The electron micrographs at higher magnitudes (Figs 3b and c) showed that some adjacent particles fused with one another. No diffraction spots or rings for an individual particle were observed, indicating that these particles were amorphous. Nanosheets started to appear after a reaction time of 5 h (Fig. 3d). Upon further prolonging the reaction time to 15 h, most of the spherical structures were transformed into nanosheets (Fig. 3e). After 24 h, only nanosheets were found, with a relatively broad size distribution (Fig. 3f).
 | ||
| Fig. 3 SEM (a and b) and TEM (c) images of the spherical nanostructures prepared with complex 1 after a mixing time of 1 h. SEM images depicting the morphological evolution from spherical particles to nanosheets after a mixing time of 5 (d), 15 (e) and 24 (f) hours. | ||
To gain an insight into the growth mechanism of these nanosheets, the solvent dependence of the transformation from particles to sheet-like structures was examined. The proportion of dichloromethane in the mixed solvent was found to significantly impact on the growth rate of the nanosheets. For example, when 3 mL of n-hexane was added to a solution of complex 1 in 200 μL of dichloromethane, the particles were completely transformed to nanosheets within 5 min. In contrast, the transformation process took more than 15 h if only 100 μL of dichloromethane was used. Furthermore, removal of dichloromethane from the suspensions was found to be crucial to prevent the generation of nanosheets. When the particles were collected by repeated centrifugation and re-dispersed in hexane (the poor solvent), no nanosheets were found even after a few weeks. Similar transformations from spherical objects to sheet-like structures have also been observed when acetone/hexane was used as the solvent system. Based on these results, we suggest that a dynamic recrystallization process of the copper(I) complexes in solvent mixtures of good (dichloromethane or acetone) and poor (hexane) solvents is responsible for the formation of nanosheets from spherical particles. This morphological and crystalline evolution is similar to the shape transformation from nanowires to nanocubes observed with coordination polymer particles,13 but is somewhat different from the fabrication of nanotubes through the 1D fusion of nanocapsules.14
Photophysical properties
Complexes 1 and 2 in dichloromethane exhibit an intense absorption at 250–550 nm with a large extinction coefficient (∼104 mol−1 dm3 cm−1). The absorption bands in the region of 250–400 nm originate from 1π–π* transitions localized on the naphthyridine ligand. At this stage, the low-energy absorptions in the 400–500 nm region are tentatively assigned to 3d(Cu)→π*(L) 1MLCT transitions, with reference to previous works on [Cu(NN)2]+ (NN = diimine ligands) systems.6a The emission peaks of complexes 1 and 2 in degassed dichloromethane are broad, with λmax (lifetime, quantum yield) of 602 nm (0.11 μs, 1%) and 613 (0.32 μs, 2%), respectively. Lowering the temperature from 298 K to 77 K resulted in a blue shift in the emission energy, together with a dramatic increase in the emission lifetime up to 155 μs for 1 and 267 μs for 2. Crystalline samples of 1 and 2 display emissions with a λmax (lifetime) of 565 nm (0.25 μs) and 609 nm (0.35 μs), respectively, both of which do not change significantly in emission energy upon cooling to 77 K (570 nm, 60.4 μs; 612 nm, 112.8 μs, respectively). It should be emphasized that the emissions of 1 in solution and in the solid state are different in energy, while those of 2 in solution and in the solid state display almost the same emission λmax wavelength.Nanostructures of complex 1 were found to display morphology/crystallinity-dependent phosphorescence. For example, orange (λmax = 600 nm) and greenish yellow (λmax = 560 nm) emissions were observed for the amorphous particles and crystalline nanosheets prepared with complex 1, respectively (Fig. 4c). The spectral blue-shift from amorphous to crystalline phases, together with a remarkable enhancement in the emission intensity is unusual, because the crystallization of dye molecules in the solid state generally red-shifts and weakens their emissions.15 We note that the emissions of the spherical particles and nanosheets prepared with complex 1 are close in energy to those in molecular solutions (λmax = 602 nm) and in crystalline samples (λmax = 565 nm), respectively. This implies that the excited-state structures of 1 in solutions and in amorphous particles are similar. In contrast, the emission maxima of complex 2 in solution (613 nm) and in solid state (609 nm) are at a similar wavelength, which accounts for the lack of blue-shift in the emission energy upon crystallization.
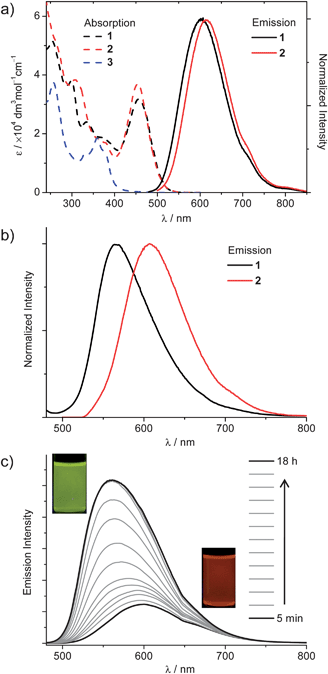 | ||
| Fig. 4 (a) Absorption spectra of 1–3 and emission spectra of 1–2 in dichloromethane. (b) Emission spectra of 1 and 2 in the solid state at room temperature. (c) Emission traces of a dispersion of complex 1 as a function of aging time after the addition of 3 mL of n-hexane to a dichloromethane solution of 1 (∼1.5 mM, 0.1 mL), λex = 420 nm. | ||
Density functional theory calculations
In order to understand the differences in the emissive behaviour of complexes 1 and 2 in solution and as crystalline nanostructures, Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT) were employed for geometry optimizations of the singlet ground state (S0) and the lowest triplet excited state (T1) and the calculations of the excited states at the optimized geometries, respectively (see ESI† for details). For both complexes, the four lowest-lying singlet excited states at λ > 420 nm are derived from HOMO/H − 1 → LUMO/L + 1 transitions where the HOMO/H − 1 and LUMO/L + 1 are quasi-degenerate pairs. The HOMO and H − 1 orbitals are mainly composed of the two central Cu(I) ions (Cu1 and Cu1′, Fig. 5a) and π-orbitals delocalized over the heterocyclic ligand L (HL = N,N′-bis(5,7-dimethyl-1,8-naphthyridine-2-yl)amine), i.e., π(Np1/N1–amido/Np2) and π(Np3/N1′–amido/Np4)). The LUMO and L + 1 orbitals are made up of π*(Np1/Np2) and π*(Np3/Np4). The pairs differ only in phase with respect to the inversion centre of the complex (both complexes have pseudo-Cs symmetry).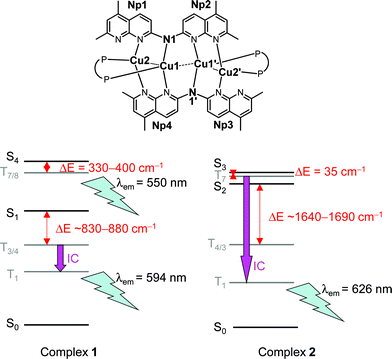 | ||
| Fig. 5 (a) Labeling of the structures of complexes 1 and 2 involved in the theoretical calculations. (b) Energy diagrams and photophysical pathways of complexes 1 and 2 in their optimized geometries of ground and triplet excited states (TDDFT results). | ||
For complex 1 (Fig. 5b, left), the allowed singlet excited states are S1 and S4, and there are close-lying triplet excited states for facile intersystem crossing (ISC) from these two singlet excited states (ΔE (S1−T3/4) = 880 / 830 cm−1 and ΔE (S4−T7/8) = 400 / 330 cm−1, see ESI†). Thus, at λex = 420 nm, both the S1 and S4 excited states of complex 1 could be populated and undergo facile ISC to different triplet excited states. In dichloromethane solution or in the aggregate state, the large entropy may favour the population of the lowest singlet excited state S1 such that there is fast internal conversion from S4 to S1 after photo-excitation, followed by efficient ISC from S1 to T3/4 and fast internal conversion to the T1 excited state. The T1 excited state was calculated to be at λ = 594 nm, comparable to solution emission maximum at λ = 602 nm observed experimentally. However, in the crystalline state, it is enthalpy favoured as the atom movements are more constrained and the population of the S4 excited state increases. This S4 excited state could also undergo fast ISC to T7/8 excited states (vide supra), where fast internal conversion to the T1 excited state may be suppressed as the complex is now in the crystalline state and thus emission takes place at higher-lying triplet excited states. It is estimated that the emission at this higher-lying triplet excited state would be at ~550 nm, which is in good agreement with the emission maximum at 565 nm in the crystalline state. As the atom movements are suppressed in the crystalline state, non-radiative decay is slower than that in solution. Hence, the quantum yield of this higher-lying triplet excited state is larger.
On the other hand, for complex 2 (Fig. 5b, right), the allowed singlet excited states are S2 and S3. Here, only the S3 excited state has a close-lying triplet excited state for ISC (ΔE (S3−T7) = 35 cm−1); the closest-lying triplet excited state for S2 to undergo ISC is more than 1600 cm−1 (see ESI†). As such, after photoexcitation at λ = 420 nm, only the S3 excited state would undergo facile ISC and hence, there is only one emissive state for complex 2. Complex 2 has different excited state parentages from those of complex 1, most likely due to the presence of the phenyl groups in the phosphine ligands. In order to have π-π interactions between the phenyl rings on the phosphine ligand and the Np2 and Np4 rings of ligand L, the angles N–Cu–N is larger for complex 2 than those for complex 1. In effect, the occupied orbital with gerade symmetry is higher in energy than the orbital with ungerade symmetry for complex 2 (see ESI†). As complex 2 is less densely packed than complex 1 in the crystalline state, the former may undergo fast internal conversion to the T1 excited state whether it is in solution or in the crystalline state. The emission from T1 excited state was calculated to be at 626 nm, comparable to the experimental observed emission maximum at 613 nm. Again, the increase in emission quantum yield may possibly be due to the slower non-radiative decay in the solid state.
Conclusion
In conclusion, low-molecular-weight tetranuclear copper(I) complexes with phosphorescence and fluxional behaviors can assemble into crystalline quasi-2D sheet-like nanostructures, with amorphous spherical particles as an intermediate state. The morphological evolution and crystallization-induced emission enhancement from particles to nanosheets were studied. Both the experimental data and DFT calculations indicate that a subtle variation of the ligand system on the tetranuclear copper(I) complexes has a profound effect on the excited state geometries and, consequently, the photophysical pathways in solutions, in molecular aggregates and in crystalline solids. This work may stimulate further investigations into the structure–morphology–property relationships of nanostructures assembled from luminescent transition metal complexes.Acknowledgements
This work was supported by the foundation (GJHZ200817) for Bureau of International Co-operation of CAS. We thank the National Basic Research Program of China (973 program 2007CB613304) and the National Natural Science Foundation of China (No. 21071123 and 21001110) for financial support. This work in Hong Kong was supported by the University Development Fund of The University of Hong Kong and the Hong Kong Research Grants Council (HKU 7008/09P and AoE/P-03/08), NSFC/RGC Joint Research Scheme (N_HKU 752/08), and CAS-Croucher Funding Scheme for Joint Laboratories. The calculations in this project are supported by a Hong Kong UGC Special Equipment Grant (SEG HKU09). Thanks go to Mr. Frankie Yu-Fee Chan and Ms. Amy Sui-Ling Wong at HKU for assistance in electron microscopy.Notes and references
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- G. D. Scholes and G. Rumbles, Nat. Mater., 2006, 5, 683–696 CrossRef CAS.
- (a) J. J. Chiu, C. C. Kei, T. P. Perng and W. S. Wang, Adv. Mater., 2003, 15, 1361–1363 CrossRef CAS; (b) Y. S. Zhao, C. A. Di, W. S. Yang, G. Yu, Y. Q. Liu and J. N. Yao, Adv. Funct. Mater., 2006, 16, 1985–1991 CrossRef CAS; (c) J. S. Hu, H. X. Ji, A. M. Cao, Z. X. Huang, Y. Zhang, L. J. Wan, A. D. Xia, D. P. Yu, X. M. Meng and S. T. Lee, Chem. Commun., 2007, 3083–3085 RSC; (d) W. Chen, Q. Peng and Y. Li, Adv. Mater., 2008, 20, 2747–2750 CrossRef CAS.
- (a) Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS; (b) Z. Wang, Z. Li, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2007, 129, 2440–2441 CrossRef CAS; (c) J. S. Hu, Y. G. Guo, H. P. Liang, L. J. Wan and L. Jiang, J. Am. Chem. Soc., 2005, 127, 17090–17095 CrossRef CAS; (d) Z. Wang, K. J. Ho, C. J. Medforth and J. A. Shelnutt, Adv. Mater., 2006, 18, 2557–2560 CrossRef CAS; (e) M. H. So, V. A. L. Roy, Z. X. Xu, S. S. Y. Chui, M. Y. Yuen, C. M. Ho and C. M. Che, Chem.–Asian J., 2008, 3, 1968–1978 Search PubMed; (f) C. J. Medforth, Z. Wang, K. E. Martin, Y. Song, J. L. Jacobsen and J. A. Shelnutt, Chem. Commun., 2009, 7261–7277 RSC.
- (a) W. Lu, V. A. L. Roy and C. M. Che, Chem. Commun., 2006, 3972–3974 RSC; (b) Y. H. Sun, K. Q. Ye, H. Y. Zhang, J. H. Zhang, L. Zhao, B. Li, G. D. Yang, B. Yang, Y. Wang, S. W. Lai and C. M. Che, Angew. Chem., Int. Ed., 2006, 45, 5610–5613 CrossRef CAS; (c) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J. P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659–2662 CrossRef CAS; (d) A. Y. Y. Tam, K. M. C. Wong, G. X. Wang and V. W. W. Yam, Chem. Commun., 2007, 2028–2030 RSC; (e) W. Lu, S. S. Y. Chui, K. M. Ng and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568–4572 CrossRef CAS; (f) M. Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M. H. So, S. S. Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895–9899 CrossRef CAS; (g) W. Lu, K. M. Ng and C. M. Che, Chem.–Asian J., 2009, 4, 830–834 CrossRef CAS; (h) W. Lu, Y. Chen, V. A. L. Roy, S. S. Y. Chui and C. M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621–7625 CrossRef CAS; (i) Y. Chen, K. Li, W. Lu, S. S. Y. Chui, C. W. Ma and C. M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS; (j) K. Jang, I. G. Jung, H. J. Nam, D.-Y. Jung and S. U. Son, J. Am. Chem. Soc., 2009, 131, 12046–12047 CrossRef CAS; (k) Y. Chen, K. Li, H. O. Lloyd, W. Lu, S. S. Y. Chui and C. M. Che, Angew. Chem. Int. Ed. 2010499968–9971 Search PubMed.
- For recent reviews on copper(I) complexes, see: (a) D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1220 CrossRef; (b) P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3647 CrossRef CAS; (c) V. W. W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC; (d) D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS; (e) N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC; (f) N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CAS.
- (a) Y. G. Ma, W. H. Chan, X. M. Zhou and C. M. Che, New J. Chem., 1999, 23, 263–265 RSC; (b) Y. Ma, C. M. Che, H. Y. Chao, X. Zhou, W. H. Chan and J. Shen, Adv. Mater., 1999, 11, 852–857 CrossRef CAS; (c) Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Mater., 2004, 16, 432–436 CrossRef CAS; (d) J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS.
- (a) W. H. Chan, S. M. Peng and C. M. Che, J. Chem. Soc., Dalton Trans., 1998, 2867–2871 RSC; (b) C. M. Che, Z. Mao, V. M. Miskowski, M. C. Tse, C. K. Chan, K. K. Cheung, D. L. Phillips and K. H. Leung, Angew. Chem., Int. Ed., 2000, 39, 4084–4088 CrossRef CAS; (c) Z. Mao, H. Y. Chao, Z. Hui, C. M. Che, W. F. Fu, K. K. Cheung and N. Zhu, Chem.–Eur. J., 2003, 9, 2885–2894 CrossRef CAS; (d) W. F. Fu, X. Gan, C. M. Che, Q. Y. Cao, Z. Y. Zhou and N. Zhu, Chem.–Eur. J., 2004, 10, 2228–2236 CrossRef CAS; (e) Y. Chen, J. S. Chen, X. Gan and W. F. Fu, Inorg. Chim. Acta, 2009, 362, 2492–2498 CrossRef CAS.
- (a) Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H.-Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC; (b) Y. Dong, J. W. Y. Lam, A. Qin, J. Sun, J. Liu, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Chem. Commun., 2007, 3255–3257 RSC; (c) L. Qian, B. Tong, J. Shen, J. Shi, J. Zhi, Y. Dong, F. Yang, Y. Dong, J. W. Y. Lam, Y. Liu and B. Z. Tang, J. Phys. Chem. B, 2009, 113, 9098–9013 Search PubMed; (d) C. Park, E. Yoon, M. Kawano, T. Joo and H. C. Choi, Angew. Chem., Int. Ed., 2010, 49, 9670–9675 CrossRef CAS.
- Y. Chen, W. F. Fu, J. L. Li, X. J. Zhao and X. M. Ou, New J. Chem., 2007, 31, 1785–1788 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) H. Schmidbaur and K. C. Dash, J. Am. Chem. Soc., 1973, 95, 4855–4860 CrossRef CAS; (b) K. Dixon, Inorg. Chem., 1977, 16, 2618–2624 CrossRef CAS; (c) J. B. Brandon, M. Collins and K. Dixon, Can. J. Chem., 1978, 56, 950–953 Search PubMed; (d) C. Bianchini, H. M. Lee, P. Barbaro, A. Meli, S. Moneti and F. Vizza, New J. Chem., 1999, 23, 929–938 RSC; (e) T. Koizumi and K. Tanaka, Inorg. Chim. Acta, 2004, 357, 3666–3672 CrossRef CAS; (f) T. Suzuki, Inorg. Chim. Acta, 2006, 359, 2431–2438 Search PubMed.
- S. Jung and M. Oh, Angew. Chem., Int. Ed., 2008, 47, 2049–2051 CrossRef CAS.
- K. Kondo, T. Kida, Y. Ogawa, Y. Arikawa and M. Akashi, J. Am. Chem. Soc., 2010, 132, 8236–8237 Search PubMed.
- (a) A. B. Koren, M. D. Curtis and J. W. Kampf, Chem. Mater., 2000, 12, 1519–1522 CrossRef CAS; (b) A. B. Koren, M. D. Curtis, A. H. Francis and J. W. Kampf, J. Am. Chem. Soc., 2003, 125, 5040–5050 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, supplementary photophysical data and electron micrographs. CCDC reference numbers 793442–793444. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00597e |
‡ CCDC 793442–793444 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif. Crystal data. 1·CH2Cl2: C91H130B2Cl2Cu4F8N10P4, M = 1986.61, monoclinic, a = 27.568(7), b = 15.989(4), c = 25.240(6) Å, β = 106.726(5)°. V = 10655(5) Å3, T = 293(2)K, space groupC2/c, Z = 4, 27424 reflections measured, 9375 unique (Rint = 0.0820), which were used in all calculations. The final wR(F2) was 0.2251 (all data). 2·2CH2Cl2: C92H84B2Cl4Cu4F8N10P4, M = 2023.15, monoclinic, a = 12.238(2), b = 18.531(3), c = 20.929(3) Å, β = 103.246(3)°. V = 4620.2(1) Å3, T = 298(2)K, space groupP21/n, Z = 2, 23854 reflections measured, 8143 unique (Rint = 0.0477), which were used in all calculations. The final wR(F2) was 0.1142 (all data). 3: C38H34BCuF4N5P, M = 742.02, triclinic, a = 8.485(2), b = 13.769(2), c = 15.231(3) Å, α = 91.110(2)°, β = 100.453(3)°, γ = 91.010(2)°. V = 1749.2(6) Å3, T = 293(2)K, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 9117 reflections measured, 6063 unique (Rint = 0.0513), which were used in all calculations. The final wR(F2) was 0.1270 (all data). , Z = 2, 9117 reflections measured, 6063 unique (Rint = 0.0513), which were used in all calculations. The final wR(F2) was 0.1270 (all data). |
| This journal is © The Royal Society of Chemistry 2011 |