Evaluation of an immobilized artificial carbonic anhydrase model for CO2 sequestration†
Lan-Ya
Cheng
a,
Yi-Tao
Long
*a,
Heinz-Bernhard
Kraatz
*b and
He
Tian
a
aShanghai Key Laboratory of Functional Materials Chemistry & Institute of Fine Chemicals, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: ytlong@ecust.edu.cn; Fax: +(86)-21-64252339; Tel: +(86)-21-64252339
bDepartment of Chemistry, The University of Western Ontario, London, Ontario N6A 5B7, Canada. E-mail: hkraatz@uwo.ca; Fax: +(519)661-3022 x81561; Tel: +(519)661-3022 x81561
First published on 19th May 2011
Abstract
An immobilized artificial carbonic anhydrase model for CO2 sequestration was established by immobilizing a novel Fc-histidine conjugate on gold surfaces and then chelating with Zn2+ ions. Its catalytic activity was investigated using electrochemical and spectroscopic methods, respectively.
Carbon dioxide (CO2) sequestration has attracted considerable interest due to its potential contribution to a low carbon economy and removal/storage of CO2 gas.1 Particular attention has been paid to CO2 biocatalytic or biomimetic convertion, especially through carbonic anhydrase (CA, a Zn2+ containing enzyme in Nature) and its analogues, which can catalyze the fast reversible conversion between HCO3− and CO2.2 However, some disadvantages using the free CAs in solution such as the difficulty in product separation and enzymatic regeneration as well as the low efficiency in enzymatic reutilization and enzymatic stability, have become a confronted challenge. Therefore, various immobilized CAs are investigated to eliminate the disadvantages of free CAs.3 To the best of our knowledge, no work on the immobilized CA analogues is reported so far. Here we demonstrate an artificial CA model on gold surface and evaluate its catalytic activity.
The core design of an artificial CA model is to construct a structure which can simulate the active site of the natural CA enzyme, that is, three histidine imidazoles coordinating to a Zn2+ ion with a water molecule completing the tetrahedral coordination environment of the metal center.4 In order to obtain the expected structure, a prepared Fc-histidine conjugate (FcSSHG) with two His(DNP)-Gly-Ome peptide sequences and a cystamine moiety, was immobilized on the gold surface and consequently chelated with Zn2+ ions. As a result, the self-assembled monolayer of FcSSHG (SAM-Fc) and its Zn artificial model (Zn-AM) were formed (ESI†). The Fc group is enrolled as the electrochemical indicator to evaluate the property of the film. The cystamine moiety affords to attach the FcSSHG molecules with gold through its sulfur atoms.5 The peptide sequence His(DNP)-Gly-Ome is employed to supply Zn2+ ions with the binding sites, since it has been proved in earlier reports that this peptide sequence is able to form stable coordination with Zn2+ ions through the imidazole N (Nimi) atoms of histidines.6 We envisioned that a Zn2+ ion coordination binding with the Nimi atoms can be formed in the Zn-AM. A quasi-tetrahedral configuration including three Nimi atoms and one Zn2+ ion is expected to form as the active center in the Zn-AM, seen as Fig. 1a.
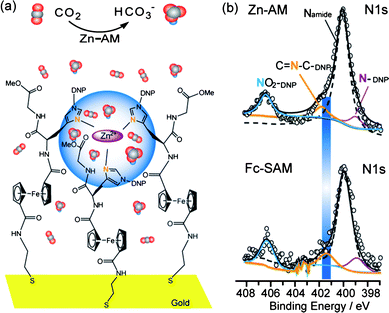 | ||
| Fig. 1 (a) The expected schematic representation of CO2 hydration catalyzed by Zn-AM on gold electrode. The blue area indicates the active center. The line-model represents the CO2 molecule, and the triangle-model represents the HCO3− ion. (b) Comparison between the detailed N1s spectra for SAM-Fc and Zn-AM on gold substrate. Open circles stand for experimental raw data, black solid lines for the total fit, orange solid lines for the fitted peaks of the free Nimi, purple solid lines for the fitted peaks of the Nimi protected by DNP, cyan solid lines for the fitted peaks of NO2 on DNP and the black dash lines for the fitted peaks of amide N. The blue rectangle emphasized the shift of the free Nimi between SAM-Fc and Zn-AM. | ||
First, we confirmed the formation of SAM-Fc and Zn-AM using electrochemical techniques and X-ray Photoelectron Spectroscopy (XPS). As seen in Figure S4–5,† a typical redox signal for the Fc/Fc+ couple is observed at 635mVvs.SCE. in the cyclic voltammetry (CV) plot of SAM-Fc. Moreover, a good linear relation is also displayed between its anodic current ip and scan rate, which suggests a definite surface controlled electrochemical activity. Based on the gold electrode area and the charge associated with Fc/Fc+ redox couple,7 the surface concentration of Fc (ΓFc) and the corresponding footprint area were calculated as 6.92 × 10−11 mol cm−2 and 240 Å2, respectively. These values are consistent with other Fc-peptide monolayer films, indicating that the Fc-SAM existed on the gold surface approximately as monolayer.5b, 8
In the detailed XPS spectra of SAM-Fc (Figure S8a†), the S2p signals appeared at the binding energy of 163.3 eV and 162.1 eV, which indicates that the Fc-SAM attached the gold surface through Au-S bond.9 The CV plot of Zn-AM (Figure S6†) shows distinct current reduction relative to that of SAM-Fc, strongly implying that Zn2+ ions have chelated on the SAM-Fc.10 The XPS signals at the binding energy of 1040.8 eV and 1017.8 eV (Figure S8b†) are assigned to Zn 2p1/2 and Zn 2p3/2,11 indicating the existence of Zn2+ ions in the Zn-AM. Moreover, the detailed XPS spectra of N1s and O1s are concerned in both of the SAM-Fc and the Zn-AM, since the N and O atoms are the potential sites to coordinate with Zn2+ ions. It is found that no obvious difference occurs on the O1s spectra, while the binding energy at 401.4 eV assigned to the Nimi1s in the SAM-Fc shifts to 401.8 eV in the Zn-AM (Fig. 1b),12 which strongly suggests that the Zn2+ ions chelated with the Nimi atoms in the Zn-AM. The matching ratio between the Nimi and the Zn atoms, equal to the surface concentration ratio between Fe and Zn atoms in the Zn-AM, is obtained as 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, according to the quantitative analysis of the XPS spectrums for Fe2p and Zn2p in the Zn-AM (ESI†). The matching ratio is close to the ideal ratio of 3:1 in the active center, which implies a combination among three Nimi atoms and one Zn2+ ion existing in the Zn-AM, probably as a quasi-tetrahedral.
:1, according to the quantitative analysis of the XPS spectrums for Fe2p and Zn2p in the Zn-AM (ESI†). The matching ratio is close to the ideal ratio of 3:1 in the active center, which implies a combination among three Nimi atoms and one Zn2+ ion existing in the Zn-AM, probably as a quasi-tetrahedral.
Next, the catalytic property of Zn-AM was detected on CO2 gas substrate in 1 M NaClO4 solution and monitored by CV. Interestingly, the current density displayed obvious enhancement when CO2 gas was bubbled into the system with Zn-AM (Fig. 2a). According to the catalysis mechanism of CA reported in a literature,13CO2 would first transform to an intermediate [(His)3Zn-OCO2H]+ in the active center, and then immediately be released as HCO3−/CO32− ions. Consequently, the accumulated HCO3−/CO32− ions around the active center diffused in various directions, and unavoidably, some HCO3−/CO32− ions penetrated into the Zn-AM layer to compensate the Fc+. Based on the ion pair theory, we can conclude that the additional compensation effect of HCO3−/CO32− would result in the increase of current density. On the other hand, the solution acidity was detected when CO2 was bubbled over 5 min. It is found that the final solution acidity with Zn-AM (pH 5.45) was greater than that without Zn-AM (pH 5.66), which indicates that more HCO3−/CO32− ions were produced with Zn-AM than without Zn-AM. All the results above suggest that the Zn-AM has catalytic ability on CO2 hydration, and the cartoon catalysis process is illustrated as Fig. 1a.
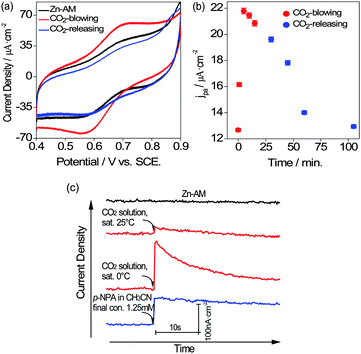 | ||
| Fig. 2 (a) The comparison of CV plots between free Zn-AM and the Zn-AM catalyzing on CO2 gas substrate in 1 M NaClO4, 25 °C. (b) Effect of CO2 gas substrate on the anodic current density of Zn-AM over time in 1 M NaClO4, 25 °C. (c) The comparison of i-t curves between free Zn-AM and the Zn-AM catalyzing various substrates. Applied potential, +0.30 V, 0.73 M NaClO4/0.09 M Tris buffer solution, pH 7.51, 25 °C. | ||
However, it was also found that the current density started to decrease when CO2 gas was kept bubbling for more than 5 min. This could be ascribed to the rapid decrease of solution acidity due to the CO2 hydration, which resulted in the inactivation of Zn-AM or the inhibition of CO2 hydration itself. Therefore a solution with a relatively stable pH condition is necessary for the catalysis of Zn-AM. To overcome the problem, a mixed tris-(hydroxymethyl) amino-methane (Tris) buffer solution (0.73 M NaClO4/0.09 M Tris) was prepared at pH 7.51 and used in the subsequent experiments. The CO2 saturated solutions were utilized instead of CO2 gas due to its easy operation and high accuracy. To carry out the successive monitoring of the catalyzed process, the amperometric i-t curve technique was employed instead of CV scanning.
Next, the catalytic property of Zn-AM was detected on CO2 saturated solution substrate in the buffer solution (0.73 M NaClO4/0.09 M Tris) and monitored by i-t curve technique. In Fig. 2c, as expected, an obvious current density enhancement of Zn-AM was observed when a 9% (v/v) CO2 saturated solution was added. The CO2 solution sample saturated at 0 °C showed a much stronger current enhancement than that saturated at 25 °C, which suggests that a higher concentration of CO2 solution would result in a stronger current signal. It is worth noting that the current density enhancement decayed rapidly with a period of ∼10 s, which was attributed to the high rate of CO2 hydration with catalyst. Hence, we tested p-nitrophenyl acetate (p-NPA) with the same system, which was usually utilized to estimate the CA activity instead of CO2 assay.14 It is obvious to find that the response time for p-NPA substrate is much longer than that for CO2 due to its much lower reaction rate.
At last, we utilized the p-NPA assay to estimate the catalytic parameters of Zn-AM. The pH condition was first optimized due to its obvious effect on the enzyme activity. Due to the fact that CA usually have catalytic activity under neutral or weak basic condition,15 as well as that CO2 sequestration was expected to perform under a mild condition,1a three 0.73 M NaClO4/0.09 M Tris buffer solutions with different pH values of 7.25, 7.51 and 7.94, were respectively prepared and utilized. The hydrolytic product, para-nitrophenol (p-NP), was monitored through the absorbance at 400 nm with Zn-AM catalysis (ESI†). In Fig. 3a, the absorbance plots of p-NPA hydrolysis were displayed at each pH condition with Zn-AM catalysis over 1800 s (The effect of p-NPA self hydrolysis has been subtracted). The plus absorbance signal demonstrated that Zn-AM catalyzed the hydrolysis of p-NPA again. The absorbance at pH 7.51 is definitely much higher than those at pH 7.25 and 7.94, indicating that the catalytic activity of Zn-AM is the highest at pH 7.51. Moreover, it is found that the activity of Zn-AM decrease faster along with the increase of the solution acidity during the recycling studies as Fig. 3b, probably due to the introduced chloride ions lowering the stability of Au-gold bond, which may result in a slow oxidation of the surface and desorption of the Fc-peptide conjugate.8b Au oxidation in the presence of Cl− was reported before.8c
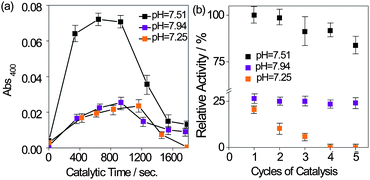 | ||
| Fig. 3 (a) p-NPA activity of Zn-AM observed through the absorbance at 400 nm in 0.73 M NaClO4/0.09 M Tris buffer solution containing 9% acetonitrile at various pH values, 25 °C. (The absorbance is calibrated by subtracting the self hydrolysis effect of p-NPA). (b) Effect of recycling on the relative activity of the Zn-AM at various pH values over 180s. The initial activity of Zn-AM at pH 7.51 is assumed as 100%. | ||
The kinetic parameters of Zn-AM were determined using Michaelis–Menten equation (eq. S2) and the Lineweaver–Burk plot (eq. S3) as literatures (ESI†).16 The absorbance intensity at 400 nm was recorded over 180 s with various p-NPA substrate concentrations ranging from 0 to 2.5 mM in the 0.73 M NaClO4/0.09 M Tris buffer solution at pH 7.51, respectively (Figure S10–11†). The apparent Michaelis constant, K′m, was determined to be 27.2 mM (Figure S11†), υ′max to be 11.7 × 10−9 mol s−1 cm−2, [E]T supposed as one third of the surface concentration of Fc-SAM, to be 2.31 × 10−11 mol cm−2, and the catalytic rate constant κcat obtained as 506.5 s−1. Since the kinetic constants depend on the CA types and experimental conditions, it is difficult to make an easy comparison between the present data and those in literature. Pointed out as reference, the K′m value of Zn-AM is close to that of the free human CA-II, to be 30.5 mM, and the κcat value, is around six times larger than that of free human CA-II, to be 79.6 s−1.17 The Zn-AM seems to catalyze the hydrolysis of p-NPA more effectively than the free human CA-II does. Similar increased κcat can be found in a variant of human CA-II with three mutations, V121A/V143A/T200A, which has κcat/KM as 1 × 105 M−1s−1 for p-NPA hydrolysis.18 We suggest that the Zn-AM exhibits catalytic activity more like a variant of the human CA-II, probably ascribed to the distinct peptide environment around the active center in the Zn-AM.
Additionally, the films of Zn-AM2 and Zn-AM3 with lower surface concentrations of FcSSHG are prepared and investigated (ESI†), respectively. Neither of them exhibits obvious catalytic activity for p-NPA hydrolysis, suggesting that the peptide sequences environment in the Zn-AM strongly affect the formation of the active center.
In conclusion, we have successfully established an immobilized artificial CA model, Zn-AM, by the Zn2+ coordination film of a Fc-histidine conjugate on gold surface. The Zn-AM clearly demonstrated catalytic activity on CO2 hydration and p-NPA hydrolysis. In addition, the kinetic parameters, K′m and κcat, were estimated to be 27.2 mM and 506.5 s−1, respectively. The results not only provide a novel way to effectively design CA model and deeply explore the mechanism of CA, but also expand the applied possibility of biomimetic CA model in CO2 sensor, capture or storage.
We greatly appreciate the support of Major Research Plan of the National Natural Science Foundation of China (Grant No.91027035), Shanghai Pujiang Program Grant of China (Grant NO.09PJ1403300) and The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning. This work was in part supported by NSERC (H.-B. Kraatz) and the University of Western Ontario.
Notes and references
- (a) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS; (b) T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS; (c) L. E. Barrosse-Antle and R. G. Compton, Chem. Commun., 2009, 3744–3746 RSC; (d) S. Ichikawa, T. Seki, M. Tada, Y. Iwasawa and T. Ikariya, J. Mater. Chem., 2010, 20, 3163–3165 RSC.
- (a) G. Parkin, Chem. Commun., 2000, 1971–1985 RSC; (b) G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS; (c) D. Huang and R. D. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4710 CrossRef CAS; (d) L. Cronin and P. H. Walton, Chem. Commun., 2003, 1572–1573 RSC.
- (a) E. Ozdemir, Energy Fuels, 2009, 23, 5725–5730 Search PubMed; (b) C. Prabhu, S. Wanjari, S. Gawande, S. Das, N. Labhsetwar, S. Kotwal, A. K. Puri, T. Satyanarayana and S. Rayalu, J. Mol. Catal. B: Enzym., 2009, 60, 13–21 Search PubMed.
- (a) X. Xu, A. R. Lajmi and J. W. Canary, Chem. Commun., 1998, 2701–2702 RSC; (b) G. Frison and G. Ohanessian, Phys. Chem. Chem. Phys., 2009, 11, 374–383 RSC.
- (a) G. A. Orlowsiki, S. Chowdhury, Y.-T. Long, T. C. Sutherland and H.-B. Kraatz, Chem. Commun., 2005, 1330–1332 RSC; (b) G. A. Orlowski, S. Chowdhury and H.-B. Kraatz, Langmuir, 2007, 23, 12765–12770 CrossRef CAS.
- (a) L.-Y. Cheng, Y.-T. Long, H. Tian and H.-B. Kraatz, Eur. J. Inorg. Chem., 2010, 33, 5231–5238 Search PubMed; (b) W. Yang, D. Jaramillo, J. J. Gooding, D. B. Hibbert, R. Zhang, G. D. Willett and K. J. Fisher, Chem. Commun., 2001, 1982–1983 RSC.
- P. A. Brooksby, K. H. Anderson, A. J. Downard and A. D. Abell, Langmuir, 2010, 26, 1334–1339 Search PubMed.
- (a) F. E. Appoh and H.-B. Kraatz, J. Phys. Chem. C, 2007, 111, 4235–4245 Search PubMed; (b) G. A. Orlowski, S. Chowdhury and H.-B. Kraatz, Electrochim. Acta, 2007, 53, 2034–2039 CrossRef CAS; (c) see for example: K. Kerman and H.-B. Kraatz, Chem. Commun., 2007, 5019–5021 Search PubMed.
- O. Cavalleri, G. Gonella, S. Terreni, M. Vignolo, P. Pelori, L. Floreano, A. Morgante, M. Canepa and R. Rolandi, J. Phys.: Condens. Matter, 2004, 16, S2477–S2482 CrossRef CAS.
- (a) K. Uosaki, Y. Sato and H. Kita, Langmuir, 1991, 7, 1510 CrossRef CAS; (b) G. K. Rowe and S. E. Creager, Langmuir, 1991, 7, 2307–2312 CrossRef CAS.
- (a) Z. Peng, X. Qu and S. Dong, J. Electroanal. Chem., 2004, 563, 291–298 Search PubMed; (b) R. Andreu, J. J. Calvente, W. R. Fawcett and M. Molero, J. Phys. Chem. B, 1997, 101, 2884–2894 CrossRef CAS.
- (a) X. Bi and K.-L. Yang, Langmuir, 2007, 23, 11067–11073 Search PubMed; (b) G. Liu, Q. T. Nguyen, E. Chow, T. Böcking, D. B. Hibbert and J. J. Gooding, Electroanalysis, 2006, 18, 1141–1151 CrossRef CAS.
- (a) B. C. Tripp, K. Smith and J. G. Ferry, J. Biol. Chem., 2001, 276, 48615–48618 CrossRef; (b) C. A. Morgado, I. H. Hillier, N. A. Burton and J. J. W. McDouall, Phys. Chem. Chem. Phys., 2008, 10, 2706–2714 RSC.
- R. G. Khalifah and D. N. Silverman, The Carbonic Anhydrases: Cellular Physiology and Molecular Genetics, S. J. Dodgson, R. E. Tashian, G. Gros and N. D. Carter, ed., Plenum Press: New York, 1991, 49–70 Search PubMed.
- S. Toba, G. Colombo and K. M. Merz, J. Am. Chem. Soc., 1999, 121, 2290–2302 CrossRef CAS.
- (a) A. Ramundo-Orlando, G. P. Gallerano, P. Stano, A. Doria, E. Giovenale, G. Messina, M. Capelli, M. D’Arienzo and I. Spassovsky, Bioelectromagnetics, 2007, 28, 587–598 Search PubMed; (b) A. Ramundo-Orlando, F. Mattia, A. Palombo and G. D’Inzeo, Bioelectromagnetics, 2000, 21, 499–507 Search PubMed.
- A. Innocenti, A. Scozzafava, S. Parkkila, L. Puccetti, G. De Simone and C. T. Supuran, Bioorg. Med. Chem. Lett., 2008, 18, 2267–2271 Search PubMed.
- G. E. Höst and B.-H. Jonsson, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 811–815 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure and characterization of FcSSHG. Preparation and Characterization of Fc-SAM and Zn-AM. Detail of p-NPA assay. See DOI: 10.1039/c1sc00028d |
| This journal is © The Royal Society of Chemistry 2011 |