Gas-phase H/D-exchange reactions on resorcinarene and pyrogallarene capsules: Proton transport through a one-dimensional Grotthuss mechanism
Henrik D. F.
Winkler
a,
Egor V.
Dzyuba
a,
Julian A. W.
Sklorz
a,
N. Kodiah
Beyeh
b,
Kari
Rissanen
b and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie and Center of Supramolecular Interactions (CSI Berlin), Freie Universität, Takustr. 3, D-14195, Berlin, Germany. E-mail: c.schalley@schalley-lab.de.; Fax: +49-30-838-55817; Tel: +49-30-838-52639
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, PL 35, FIN-40014, Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi; Fax: +358-50-562-3721; Tel: +358 -14-260-2672
First published on 7th January 2011
Abstract
Hydrogen/deuterium exchange (HDX) experiments can be used to examine the gas-phase structure of hydrogen-bonded dimeric resorcinarene and pyrogallarene capsules. Already the qualitative comparison of the isotope exchange rates of different host–guest complexes with Cs+, tetramethyl ammonium (TMA+) and tetraethyl ammonium (TEA+) as the guest cations provides insight into the H/D-exchange mechanisms and with it, into the capsules' gas-phase ion structures. The smaller Cs+cations bind inside dimeric capsules with an intact seam of hydrogen bonds between the two monomers. Larger cations such as TEA+ lead to capsules with partially disrupted seams of hydrogen bonds. A fast isotope exchange is only observed, when the H-bonding seam between the two monomers is intact. In these cases, the H/D-exchange proceeds by a concerted mechanism reminiscent of the Grotthuss mechanism of proton transfer through water. Since it can only proceed along the seam, we refer to this exchange mechanism as a one dimensional Grotthuss mechanism.
Introduction
Well controlled proton transfer plays a fundamental role for energy conversion in living cells.1 For example, ATP synthase2,3 utilizes a proton gradient to drive a directional proton translocation across the cell membrane to generate ATP which provides the cell with a source of chemical energy and fuels its metabolism. Aquaporins1,4 are membrane pores which allow water to cross the cell membrane. However, their intelligent construction prevents a simultaneous transport of water molecules andprotons through the membrane and thus keeps the proton gradients intact during the water transport through the membrane. This is achieved by an “hourglass”-shaped water channel which ensures that no continuous chain of hydrogen-bonded water molecules can connect the inside and the outside bulk water. These two examples illustrate nicely how important the details of proton transfer through water are for the cells to function.Proton transfer through water is faster and with it electric conductivity higher than expected for hydronium ions to occur. In his seminal paper dating back to 1806, de Grotthuss suggested a proton hopping mechanism as depicted in Fig. 1.5 This mechanism implies that the proton travels only virtually through water in that a sequence of short-distance proton hopping steps transports the charge over a long distance. Much research has been invested to unravel the details of this mechanism.6–11 Today, it is widely accepted that it involves the Zundel cation (H5O2+) and the Eigen cation (H9O4+) as intermediates, both representing differently solvated hydronium ions (Fig. 1). The individual proton transfer steps between two water oxygen atoms proceeds on the picosecond time scale.12 For this fast proton transport, it is essential that the free energy of both cationic transport stages lies quite close to each other and the energy barrier between them is low.13
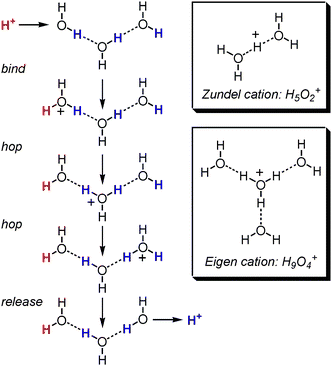 | ||
| Fig. 1 The Grotthuss mechanism of proton transport through water consists of several proton hopping steps and is believed to proceed through the Zundel and Eigen cations. | ||
One of the inspirations and main goals of supramolecular chemistry is to design and prepare simple model systems that can mimic biochemical molecules and processes in order to study their underlying principles. In the present study, we aimed at investigating the gas-phase H/D-exchange behavior of monomeric and dimeric complexes of resorcinarenes14–19 and pyrogallarenes20–22 with different guest cations (Fig. 2). One goal is to devise an experiment which would provide evidence for the closed capsular structure of the dimeric reso and pyrocapsules in the gas phase. The logic behind this is that a capsular dimer with a completely closed seam of hydrogen bonds between the two monomeric halves might exhibit an H/D-exchange behaviour different from that of a host–guest complex with a (partially) open seam. A second goal is to put H/D-exchange mechanisms to the test and to obtain a clear picture of the underlying mechanistic details.
![Structures of the resorcin[4]arene (reso) and the pyrogall[4]arene (pyro) monomer. Tetramethylated resorcin[4]arene (reso-Me) serves as a control compound. The inset shows the cations investigated as the guests for the monomers and reso and pyro dimers. Labile OH hydrogen atoms are marked in blue.](/image/article/2011/SC/c0sc00539h/c0sc00539h-f2.gif) | ||
| Fig. 2 Structures of the resorcin[4]arene (reso) and the pyrogall[4]arene (pyro) monomer. Tetramethylated resorcin[4]arene (reso-Me) serves as a control compound. The inset shows the cations investigated as the guests for the monomers and reso and pyro dimers. Labile OH hydrogen atoms are marked in blue. | ||
Resorcinarenes and pyrogallarenes are able to form a wide variety of host–guest complexes.14,16,23–26 Their three-dimensional bowl-shaped cavity is stabilized by four intramolecular OH⋯O hydrogen bonds as shown in Fig. 2. It consists of four electron-rich aromatic rings that offer multiple binding sites with which guests can interact for example through C–H⋯π and cation⋯π interactions.27–33 Additionally, OH groups on the upper rim of the bowl pointing away from the cavity can form hydrogen bonds with suitable guest molecules.34 As the three dimensional structure of resorcinarenes and pyrogallarenes relies on more or less weak non-covalent interactions, the investigation of structure and reactivity is often challenging. Electrospray ionization (ESI) mass spectrometry is able to transfer the molecules together with their guests into the highly diluted gas-phase inside a mass spectrometer35,36 where the intrinsic properties of those weak bonds can be studied without any interfering effects introduced by the environment. Nevertheless, mass spectrometry only provides indirect information about the structure of non-covalent ions in the gas phase.37–39 New experimental approaches that provide additional information are therefore always helpful.
HDX experiments40 in the gas phase can—within limits41—be structure indicative42 and can inter alia be utilized to study the presence of different, non- or slowly interconverting structures in an ion population as they often lead to bimodal exchange behaviour.43,44 Different hydrogen-bonding motifs usually result in different H/D exchange rates,45–47 and often hydrogen bonding can make HDX reactions very slow. Particularly striking examples are 18-crown-6 ammonium complexes, in which the crown ether acts as a non-covalent protective group for primary ammonium ions.48 Complexation to the crown makes HDX reactions on the ammonium ions several orders of magnitude slower. Beyond structure assignments, gas-phase HDX experiments also provide insight into intramolecular rearrangements within supramolecular aggregates. For example, the rapid movement of crown ethers along an oligolysine scaffold49 or on the surface of POPAM dendrimers50 was recently demonstrated by us.
For an unambiguous interpretation of the data obtained from HDX experiments, an understanding of the underlying exchange mechanism is essential.51,52 The simplest mechanism for gas-phase HDX reactions between a substrate cation and a deuteration reagent such as ND3, CH3OD, D2O, or CH3COOD consists of five consecutive, reversible steps.53,54 At first, the two reaction partners form a non-covalent encounter complex. A proton is subsequently transferred from the substrate to the reagent. Then, isotope scrambling takes place followed by back-transfer of the deuterium atom and complex dissociation into the deuterated substrate and the protonated reagent. Studies with a large range of substrates and deuteration reagents revealed the rate of H/D exchange to be inversely proportional to the difference in proton affinities of the two reaction partners. Small proton affinity differences make the proton-transfer step energetically more easily accessible. Larger differences, in particular those larger than ca. 85 kJ mol−1 usually prevent the exchange completely.53–56 Exceptions to this are amino acids or peptides, where fast HDX reactions have been observed for proton affinity differences up to 200 kJ mol−1.57 Two different mechanisms were postulated to explain these results. The exchange with more basic deuterium donors such as ND3 follows the “onium mechanism” in which a proton is transferred to the deuterating agent (Fig. 3).52 Less basic reagents such as D2O or MeOD rather follow the “relay-mechanism” in which two functional groups are involved in the simultaneous transfer of two protons between substrate and reagent.51,58
 | ||
| Fig. 3 Two HDX mechanisms in which a second functional group assists the isotope exchange mechanism. | ||
Vainiotalo et al. reported gas-phase HDX experiments with sodium-cationized monomeric and dimeric, capsular resorcinarene ions [reso·Na]+ and [reso2·Na]+, which were performed in the analyzer cell of a Fourier-transform ion-cyclotron-resonance (FTICR) mass spectrometer with ND3 as the deuteron source.59 Very surprisingly, the monomeric sodium complex did not show significant isotope exchange, while the dimeric capsule exchanges eight protons quickly followed by eight slow exchanges. An internal “flip-flop” binding motion as known from saccharide chemistry60,61 was invoked to explain, why no OH groups participated in the exchange for the monomer. In the dimeric capsule, two different sets of OH groups exist: Eight intramolecular OH⋯O hydrogen bonds stabilize the bowl conformation of the two monomers, the other eight intermolecular OH⋯O interactions mediate binding between the two monomers. While this explains the two different exchange rates observed for the dimer, it remained unclear why the dimer undergoes an HDX reaction at all. All OH hydrogen atoms are involved in hydrogen bonding and thus would not be expected to undergo any fast exchange.62 Here, we extend these studies to the gas-phase HDX on reso and pyro monomers and dimers with Cs+ and the alkylammonium ions TMA+ and TEA+. The results obtained lead to a different mechanistic interpretation in terms of a one-dimensional Grotthuss-type mechanism that explains the H/D-exchange observed in the dimeric complexes.
Experimental section
Resorcinarene (reso) was supplied by Sigma-Aldrich, Germany. The synthesis of pyro19,20,63–65 and reso-Me66 followed well-established literature procedures. All guest salts, i.e.Na2CO3, Cs2CO3, (TMA)BF4 and (TEA)BF4 and solvents were also commercially available (Sigma-Aldrich).Sample solutions for mass spectrometric experiments were prepared in acetonitrile with ∼50 μM analyte concentrations; reso, pyro or reso-Me and the guest salts were added in equimolar ratios. For this study, we have used HDX reactions in the high vacuum inside the hexapole collision cell of a 7 T Ionspec/Varian QFT7 FTICR-MS as described in detail elsewhere.49,50 The comparably high pressure (∼10−5 mbar) inside the hexapole collision cell renders the exchange more efficient as compared to that inside the FTICR analyzer cell (∼10−7 mbar).67 A second advantage is that the exchange can be done simultaneously on monomer and dimer ions under exactly the same conditions. Since monoisotopic mass-selection is not possible in the hexapole collision cell, isotope pattern deconvolution would be necessary to determine exchange rate constants. Also, the exact pressure cannot be determined so that the rate constants would be prone to larger errors. Consequently, we restrain our investigation to a qualitative evaluation of the spectra. Sample solutions containing the Cs+ ion (m/z = 133.9054) and the tetraethyl ammonium ions (m/z = 130.1596) were measured independently in order to avoid strong overlap of isotope distributions; for all measurements, the same instrumental settings and experimental conditions were used to ensure comparability. In some spectra, residual Cs+ caused memory effects and ammonium and caesium complexes overlap. However, the large difference in mass defect between the TEA+ and the Cs+ ion of ca. 0.25 amu leads to a base-line separation of the signals corresponding to both complexes. Unambiguous signal assignment and spectra evaluation is thus straightforward. The memory effect is even useful, because the HDX behaviour of the Cs+ complexes can be evaluated and compared to those obtained for them independently to make sure that comparable conditions have been used in both experiments. Reaction times between one and 1000 s were applied and deuterated methanol-d1 (CH3OD, Deutero, Germany) was used as the neutral deuterating agent. In the Figures below, we include reaction times up to 10 s only. The HDX reactivity differences are clearly reflected already in these early spectra. MeOD was introduced into the high vacuum system by means of a time-controllable solenoid pulse valve. For each exchange experiment, different reaction times were chosen as indicated in the Figures in order to be able to follow different stages of the exchange reaction. After reaction with the neutral deuterating agent, the ions were transferred into the FTICR cell for detection with a standard excitation/detection protocol.
Semi-empirical calculations on the reso and pyro dimers were perfomed using the AM1 MOZYME method as implemented in the CaChe 5.0 program package (Fujitsu Ltd., Krakow/Poland) in order to determine the most favorable hydrogen-bonding patterns connecting the two monomers. The cations have not been included in the calculations since the cation-π interactions, which play a pivotal role here, are not well described by the calculation and because the focus is on the undisturbed hydrogen bonding patterns of the reso and pyro dimers.
Results
Resorcinarene/Cs+ complexes: Monomer exchanges significantly slower than dimer
We first repeated the experiments of Vainiotalo et al.59 with the sodium-cationized resorcinarene complexes to make sure that the experiments reported here are in line with these previous results even though ND3 as the exchange reagent was replaced by CH3OD and the exchange was performed in the hexapole collision cell of our instrument rather than in the FTICR analyzer cell. This is indeed the case: Within 10 s reaction time, no exchange for [reso·Na]+ was observed, while [reso2·Na]+ revealed the exchange of a significant portion of the labile OH hydrogen atoms.However, Letzel et al.68 reported two different binding modes for alkali metal ions to resorcinarenes to exist in the gas phase. While the smaller, charge-concentrated ions (Li+, Na+) bind to the harder oxygen atoms at the upper rim, larger ions (Rb+, Cs+) prefer the cation-π interaction with the softer aromatic rings within the interior of the resorcinarene. In order to avoid any interference of cation complexation at the OH groups with the isotope exchange reaction, the reso/Cs+ complexes were thus chosen for this study.
The results for the exchange reactions with [reso·Cs]+ and [reso2·Cs]+ are shown in Fig. 4. Clearly, the monomeric complex ion undergoes only a very slow HDX reaction as indicated by the slight growth of the signal at m/z 678 over the 10 s reaction time. Instead, the dimer has already undergone up to twelve exchanges with the maximum of the isotope distribution corresponding to the exchange of five OH hydrogen atoms (from here on abbreviated as “max. at 5 HDX”). In analogy to the sodium-cationized complexes, the two Cs+ complexes again reveal a significant difference in exchange rates. This difference is puzzling because the ion, in which all OH groups are involved in hydrogen bonding undergoes a faster exchange as compared to that ion bearing free OH hydrogen atoms.
 | ||
| Fig. 4 HDX reactions performed with the Cs+-cationized reso monomer and the corresponding dimeric capsule. Exchange reagent: CH3OD; reaction times are given on the right. | ||
Resorcinarene/TEA+ complexes: Both monomer and dimer exchange slowly
The TEA+cation is certainly too large to be encapsulated inside the cavity of an intact resorcinarene dimeric capsule with a fully closed hydrogen bonding seam. Nevertheless, it can interact with two resorcinarenes through cation-π interactions to yield a PacMan-like structure.35,36,69,70 The seam of hydrogen bonds is thus only partially intact. Nevertheless, the dimer/guest complex forms with sufficient intensity in the ESI ion source. One might thus ask, whether this partially open seam might change the HDX behaviour of the [reso2·TEA]+ ion. The mass spectra in Fig. 5 clearly reveal a slow exchange not only to occur for the monomer, but also for the dimer ions. Comparing the spectra in Fig. 4 and 5, the differences between the Cs+ and TEA+ dimer complexes are obvious: The [reso2·TEA]+ ion undergoes a significantly slower exchange reaction than the corresponding Cs+ complex (max. at 2 HDX after 10 s). The isotope exchange is thus clearly sensitive to the cation size, and a cation too large to be fully encapsulated reduces the exchange rate notably. | ||
| Fig. 5 HDX reactions performed with the TEA+-cationized reso monomer and the corresponding dimer. Exchange reagent: CH3OD; asterisks: memory effect (dimer/Cs+ complex). Only the spectra recorded at 0, 4 and 10 s are displayed. | ||
Pyrogallarene/Cs+ complexes: Dimer exchanges more quickly than the pyro monomer or the analogous reso dimer
The pyrogallarene bears four additional hydroxy groups at C(2) of each aromatic ring and shows a different hydrogen bonding pattern as compared to the resorcinarenes. Therefore, a comparison of the two compounds and their complexes may provide additional mechanistic insight. Fig. 6 summarizes the exchange behavior of the Cs+-cationized pyro monomer and dimer. The results are similar to those obtained for the corresponding reso complexes. The monomer undergoes only a very slow exchange as indicated by the intensity increase of the second signal in its isotope pattern. Instead, the dimer reacts again much faster (max. at 19 HDX after 10 s). The pyrogallarene dimer bears two different OH groups, 16 at C(1) and C(3) of the pyrogallol rings, eight at the C(2) carbon atoms. The exchange of more than 16 OH hydrogen atoms after 10 s indicates both types of OH groups to exhange quickly at similar rates. | ||
| Fig. 6 HDX reactions performed with the Cs+-cationized pyro monomer and the corresponding dimer. Exchange reagent: CH3OD. | ||
Pyrogallarene/TEA+ complexes: The larger guest decelerates the exchange reaction again
For the TEA+ complexes of pyro (Fig. 7), a picture emerges similar to that observed for the corresponding reso complexes. The exchange of labile hydrogen atoms in both the [pyro·TEA]+ monomer and the [pyro2·TEA]+ dimer is quite slow. In particular, the dimer exchanges its labile hydrogen atoms (max. at 6 HDX after 10 s) much slower than the corresponding Cs+ complex (max. at 19 HDX). However, if one compares the [pyro2·TEA]+ dimer (max. at 6 HDX) with [reso2·TEA]+ (max. at 2 HDX), an unexpected difference is observed. The HDX reaction on the [pyro2·TEA]+cation is significantly faster than that of [reso2·TEA]+. Consequently, the guest cation size again plays a role for the exchange rates, but in addition, the choice of host is also a non-negligeable factor. | ||
| Fig. 7 HDX reactions performed with the TEA+-cationized pyro monomer and the corresponding dimer. Exchange reagent: CH3OD; asterisks: memory effect (monomer/Cs+ complex). | ||
TMA + complexes: A cation of intermediate size
The clear-cut exchange rate differences that are obtained, when cations of different sizes are used prompted us to examine the TMA+cation as the guest. Earlier semiempirical calculations36 suggested this cation to be fully encapsulated in the reso dimer - even though the cation fills more than 55% of the cavity, a value which has been suggested to be optimal by Rebek et al.71 The higher packing coefficient can however be easily rationalized by the operation of stabilizing cation-π interactions. In contrast, crystallographic studies always resulted in the TMA+cation encapsulated in reso dimers with solvent molecules or anions extending the hydrogen bonding seam thus providing more space inside the cavity for the guest cation.35,36,72 Consequently, TMA+ would make an interesting test case with a size right at the borderline of what the capsule would be able to accomodate.As expected, both monomeric reso and pyrohost–guest complexes undergo almost no exchange within the reaction time of 10 s (Fig. 8). Most interestingly, however, the two dimeric host–guest complexes show a distinct difference. While the exchange is almost fully blocked for [reso2·TMA]+, the analogous pyrogallarene complex [pyro2·TMA]+ shows the exchange of a portion of the labile hydrogen atoms (max. at 5 HDX after 10 s). Consequently, both dimer TMA+ complexes behave in analogy to the corresponding TEA+ complexes and again, the HDX rates depend on the choice of host.
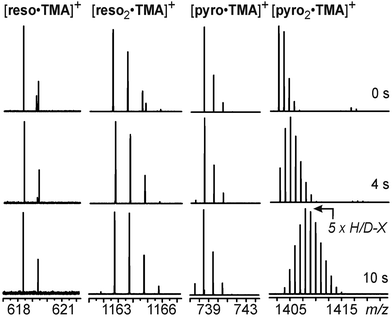 | ||
| Fig. 8 H/D-exchange mass spectra of the reso and pyro monomers and dimers with TMA+ as the guest cation. | ||
Tetramethyl resorcinarene: Control experiments with blocked hydrogen-bonding sites
Finally, tetramethyl resorcinarene reso-Me was studied as a control compound. Since it was synthesized from monomethylated resorcinol,66 the four methyl groups are arranged as shown in Fig. 2. Each aromatic ring carries one of them and all four rings carry them on the same side, because this product can form all four intramolecular OH⋯O hydrogen bonds and thus is the most favourable one. It exists in a crown conformation as the parent reso. Consequently, reso-Me is a chiral compound, but has been prepared in racemic form for this study. The four methyl groups block those OH groups which mediate the intermolecular hydrogen bonding in the unmethylated reso. Thus, reso-Me can be used as a control compound, in which the seam of hydrogen bonding in the dimer is interrupted. When mixed with reso, three dimeric capsules can be generated and compared: the reso homodimer, whose behaviour has already been discussed, the reso/reso-Me heterodimer, in which both capsule halves are connected through four hydrogen bonds, and the reso-Me homodimer, in which intermolecular hydrogen bonding does likely not occur. The latter dimer is, instead, held together by cation-π interactions between the two halves and the metal ion. It forms in the electrospray ion source with somewhat lower abundance than the other two, but can still be submitted to HDX experiments.The results from the HDX experiments conducted with the Cs+ complexes of these complexes can be briefly summarized as follows: (a) The [reso·reso-Me·Cs]+ heterodimer exchanges its labile hydrogen atoms much slower than the [reso2·Cs]+ homodimer. Its exchange rate is comparable to the [reso·Cs]+ monomer and thus the exchange becomes only visible at reaction times much longer than 10 s. Consequently, it is sufficient to partially break the hydrogen bonding seam in the reso dimer to reduce the exchange rates significantly - in line with the experiments on the TEA+ guest cations. (b) The [(reso-Me)2·Cs]+ homodimer does not show any exchange, not even after 100 s reaction time. (c) While the [reso·Cs]+ monomer undergoes a slow exchange (max. at 3 HDX after 100 s), no exchange is observed at all for the [reso-Me·Cs]+ monomer after the same reaction interval. The last two findings are particularly interesting, because they indicate that an exchange of the hydrogen atom involved in intramolecularly bridging from one resorcinol unit to the adjacent one is not directly involved in the exchange. It is the flip-flop mechanism that successively exposes the OH groups to the exchange reagent. In case of the reso-Me monomer however, this flip flop mechanism is not feasible.
Discussion
Hydrogen-bonding patterns in resorcinarene and pyrogallarene capsules: AM1 calculations
Before interpreting the HDX results in mechanistic and structural terms, it may be helpful to discuss the hydrogen-bonding patterns in the reso2 and pyro2 dimers. Many crystal structures have been reported.35,36,65,69,73,74 With one notable exception of a pyro2 dimer,65 they report capsules with an extended solvent-mediated seam of hydrogen bonds. Consequently, these studies cannot be used for comparison with the ions in the present study, because the electrospray ionization process generates fully desolvated dimer ions with a very different hydrogen-bonding pattern not interacting with any solvent molecules. As a consequence, we performed a number of simple AM1 MOZYME semiempirical calculations on different potential conformers of both dimers. The two energetically most favorable geometries for the reso2 and pyro2 dimers are shown in Fig. 9. | ||
| Fig. 9 AM1 MOZYME optimized structures of the reso (left) and pyro (right) dimers and enlargements of their H-bonding patterns (ball-and-stick representation). | ||
The two monomers in the reso2 dimer are bound in an interdigitating orientation. Each aromatic ring of one monomer dives into the space between two of the resorcinol rings of the other and vice versa. A mirror symmetrical arrangement in which the aromatic rings of one monomer directly oppose those of the other is instead not favourable. In the interdigitating structure, a hydrogen-bonding seam forms, which continuously meanders around the capsule's equator in a zigzag manner. Each OH group is hydrogen-bond donor and acceptor at the same time.
Because of the additional OH groups at the C(2) positions of the aromatic rings, the pyro2 dimer is held together by a quite different binding pattern. Again, an interdigitating structure is the most favourable one. The C(2)-OH group now participates in the formation of small, six-membered hydrogen-bonding “cells” with two adjacent C(1)- and C(3)-OH groups of the opposite monomer and vice versa. Consequently, a total of eight such “cells” are formed around the seam. This theoretical result can be directly compared to the crystal structure of a pyrogallarene dimer with an encapsulated TMA+ guest ion.65 In this solid-state structure, the two monomers are not separated by solvents mediating the hydrogen bonding, but are directly connected to each other. The calculated structure is in good agreement with the crystals structure, which confirms both, the interdigitated structure as well as the formation of the hydrogen-bonding “cells”. In addition, the crystal structure indicates the TMA+cation to be small enough to be encapsulated in a dimeric, perfectly closed pyrogallarene capsule with an intact seam of hydrogen bonds.
In order to test whether the hydrogen bonding seams would be sufficiently flexible to incorporate a MeOD molecule during the exchange reactions, methanol molecules were added to the calculated minimum-energy structures and the structures were reoptimized. In all cases, two hydrogen bonds in a reso–O–H⋯(CH3)O–H⋯O(H)-reso arrangement form between the capsule and the methanol molecule upon insertion. In both dimers, the methanol can be inserted into the seam at several non-identical sites, which have all been tested. For example, the reso2 dimer can at least in principle incorporate a methanol molecule in an intramolecular hydrogen bond. This arrangement would be different from an insertion into an intermolecular H-bond. Similarly, there are different sites in the pyro2 dimer. These calculations predict incorporation of a methanol molecule into intermolecular hydrogen bonds to be possible without disrupting the seams at any other position. The methanol molecule is mainly accommodated geometrically by OH groups turning somewhat outwards and the aromatic rings adopting a slightly more open geometry around the disturbed site. In contrast, the geometry changes are greater, the hydrogen-bond geometries less favorable and the energy penalty higher, if the methanol is to be inserted into one of the intramolecular hydrogen bonds.
Why the HDX exchange is slow for all reso monomer-guest complexes
Fig. 10 displays three mechanistic scenarios for an H/D-exchange on reso monomers. Pathway A is the classical, five-step exchange mechanism: formation of an encounter complex between reso and the MeOD exchange reagent is followed by a proton shift, isotope scrambling, a back-shift of the deuterium ion and dissociation of the complex. Mechanism B is a concerted one, which involves a six-membered transition state in which the HDX occurs. It can be seen as an analogue to the relay mechanism in Fig. 3, because both reso–OH groups actively participate. Finally, the last scenario C involves a non-concerted mechanism during which the methanol molecule is incorporated into the intramolecular hydrogen bond.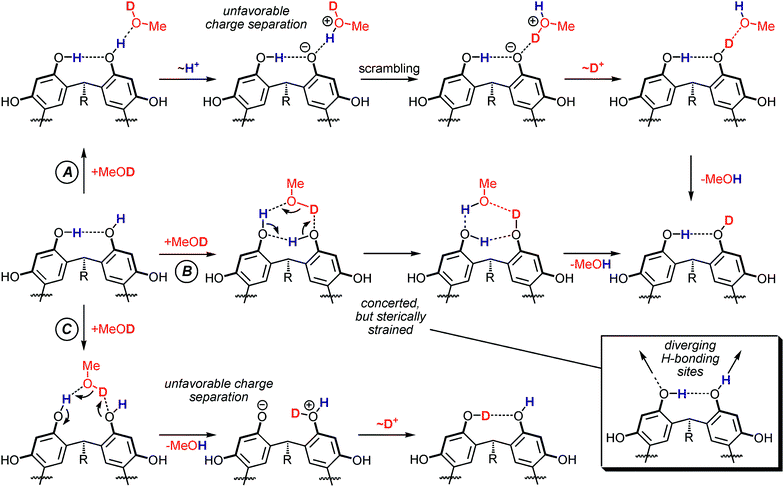 | ||
| Fig. 10 Three potential mechanistic pathways for an H/D-exchange reaction on a reso monomer. Pathway A involves only the OH group which is not involved in hydrogen bonding. B represents a concerted mechanism for the exchange proceeding through a six-membered transition state. Pathway C proceeds through an insertion of the methanol molecule into an intramolecular H-bond. | ||
Based on the arguments given above, we assume the cationic guests to be innocent spectators which do not actively participate in the exchange. The Cs+ ion likely interacts with the π-surfaces inside the bowl,68 and the ammonium ions are quaternary in nature and thus neither can donate nor accept strong hydrogen bonds. This assumption is important for the mechanistic interpretation of the HDX reaction. In contrast to the mechanisms shown in Fig. 3, the HDX reaction in the present study does not occur at the charged site, but—neglecting the innocent cation—on a neutral molecule. The consequence becomes clear from Fig. 10: In any non-concerted exchange mechanism on the reso monomer ions, an unfavorable charge separation must occur. The proton transfer steps are most favorable when the substrate and the exchange reagent have similar proton affinities. This is certainly not the case here, because the proton affinities of the phenolate-O− (1432 kJ mol−1)75 with that of methanol (754 kJ mol−1)76 are to be compared.77 Consequently, pathway A is unfavorable. Pathway C suffers from a similar problem. The energy demand for the generation of the charge-separated intermediate shown in Fig. 10 (bottom) can again be estimated based on the proton affinities of phenolate and phenol (817 kJ mol−1).76 Although this difference is somewhat smaller than that between phenolate and methanol, it is still too large to make pathway C favorable. Furthermore, the control experiments performed with tetramethylated reso-Me rule out this mechanism. Neither for [reso-Me·Cs]+ nor for the corresponding dimer [(reso-Me)2·Cs]+ any exchange occured within 400 s reaction time, while a slow exchange is indeed observed for [reso·Cs]+ in comparable time intervals. This clearly shows those OH group, which are not involved in intramolecular H-bonding, to be required for the slow exchange to occur. Consequently, the concerted mechanism B might be energetically more favourable because it avoids any charge-separation steps. However, even though it is tempting for the organic chemist to consider all six-membered transition structures favourable per se, such a transition structure is quite energy demanding here. First of all, linear OH⋯O hydrogen-bond arrangements would be preferred. This is not possible in a six-membered transition structure. In addition, the H-bond donor and acceptor sites at the upper rim of the resorcinarene diverge much as shown in Fig. 10 and in the calculated structures in Fig. 9. Therefore, such a transition state would likely be more favorable than the pathways involving charge separation, but still represents a rather unfavourable scenario. As a consequence, all three potential mechanisms in Fig. 10 are unfavorable and the observed HDX reaction is therefore very slow.
Why the HDX exchange is faster for the [reso2·Cs]+ dimer-guest complex
To explain, why the [reso2·Cs]+ dimer ion undergoes a much faster exchange reaction, we need to find a mechanism which avoids unfavorable charge separation as well as steric problems. Furthermore, the mechanism should rationalize, why the exchange of the first eight labile hydrogen atoms is faster than that of the latter eight as reported earlier for the sodium complexes.59Fig. 11 shows a concerted mechanism which is in agreement with both experimental results. After MeOD insertion into one of the intermolecular hydrogen bonds between the two capsule halves, the H/D-exchange can occur without any charge separation intermediates. A simple shift of the hydrogen atoms in all O–H⋯O towards the other oxygen yielding the corresponding O⋯H–O hydrogen bonds “short circuits” the two charges because of the complete, closed seam of hydrogen bonds. Consequently, the reaction propagates around the whole seam in a way similar to the Grotthuss mechanism for proton transport in water. The MeOD molecule simultaneously donates a deuteron on one end of the hydrogen-bonding seam and accepts a proton at the other. Since the hydrogens can only move within the seam, we define it as a one-dimensional Grotthuss mechanism.![Mercator projection of the equator region of the [reso2·Cs]+ dimer ion and an energetically favourable, concerted mechanism for H/D-exchange.](/image/article/2011/SC/c0sc00539h/c0sc00539h-f11.gif) | ||
| Fig. 11 Mercator projection of the equator region of the [reso2·Cs]+ dimer ion and an energetically favourable, concerted mechanism for H/D-exchange. | ||
Since none of the OH hydrogen atoms except for that leaving with the methanol molecule changes places with any others, it is easy to rationalize why eight fast exchanges are followed by eight slow exchanges. The eight H atoms incorporated in the intermolecular hydrogen bonds can more easily be exchanged than those involved in intramolecular hydrogen bonding and thus two different exchange rates are observed. With this mechanistic scenario, it is also easy to understand the differences in exchange rates with different cation sizes. The formation of a PacMan-shaped [reso2·TEA]+ dimer implies the hydrogen-bonding seam to be partially disrupted. Consequently, the concerted mechanism in Fig. 11 cannot operate anymore and the exchange is slowed down significantly. Finally, also the slower exchange observed for the [reso·reso-Me·Cs]+ heterodimer speaks in favor of the suggested mechanism. This dimer should be able to exist in a closed capsular form with four intermolecular hydrogen bonds connecting the two halves. However, the H-bonding seam is disrupted at the positions at which the OH groups are replaced by OMe and thus a concerted mechanism involving the whole seam is not feasible anymore.
Why the pyrogallarene dimers behave differently with the larger TMA+ and TEA+ guest cations
Overall, the exchange behaviour of the pyrogallarene complexes is similar to that of the corresponding resorcinarene complexes. However, the exchange reaction is faster for the [pyro2·TMA]+ and [pyro2·TEA]+ dimers than for the corresponding resorcinarene complexes, but slower as compared to the [pyro2·Cs]+ complex. In contrast to the reso2 dimer with its continuous H-bonding seam meandering around the capsule's equator, the pyro2 dimer possesses closed-circuit hydrogen bonding cells as discussed above. When TEA+ is bound inside a PacMan-shaped assembly, at least one of these cells will be intact and can exchange again in a concerted mechanism avoiding charge separation. In order to exchange all labile OH hydrogen atoms, a rearrangement of the PacMan would be necessary as shown in Fig. 12. This rearrangement would disrupt the cell on which the exchange has already taken place and instead close another one, where the exchange can happen next. If the rearrangement of the PacMan is the rate-determining step rather than the exchange, the apparent exchange rate would be lower than that of the reso2 or pyro2 dimers with completely closed seams. From these experiments, we conclude the Cs+-encapsulating dimers to be structurally intact capsules. The dimers with TMA+ and TEA+ in contrast appear not to have a completely closed seam of hydrogen bonds—in agreement with solution studies,35 where these complexes only appear as monomer-guest complexes and in contrast to the crystal structure65 of [pyro2·TMA+] Cl− mentioned above.![The PacMan-shaped [pyro2·TEA]+ dimer can undergo a fast exchange on one of the H-bonding “cells”. For a complete exchange of all labile hydrogen atoms, a (likely slow) rearrangement of the PacMan would however be necessary.](/image/article/2011/SC/c0sc00539h/c0sc00539h-f12.gif) | ||
| Fig. 12 The PacMan-shaped [pyro2·TEA]+ dimer can undergo a fast exchange on one of the H-bonding “cells”. For a complete exchange of all labile hydrogen atoms, a (likely slow) rearrangement of the PacMan would however be necessary. | ||
The Grotthuss analogy: A concerted mechanism?
There has been a debate whether the Grotthuss mechanism proceeds in a concerted way or not.6 Meanwhile it is becoming more and more accepted that it is very likely a stepwise process involving proton hopping. The rate-limiting step involves the cleavage of a hydrogen bond in the second solvation shell as evidenced by the activation energy of ca. 11 kJ mol−1 and a deuterium isotope effect of 1.4.From these considerations, the question arises whether the mechanism shown in Fig. 11 can be concerted at all or whether it would be more reasonable to imagine it as a very fast sequence of proton-hopping steps. Of course, our mass spectrometric experiments cannot provide a definite answer to this question, but gives some hints. First of all, any charge separation steps during the proton hopping steps would raise the energetic problems discussed above for the reso monomers, even when the intermediates exist only for very short time intervals. Consequently, a concerted mechanism is more reasonable. Furthermore, there is an important difference between water and the resorcinarene dimers. Long-range proton transport through water cannot be concerted unless long-range order of hydrogen bonded water molecules is achieved. This is certainly not the case in liquid water most likely due to entropic factors. In the resorcinarene dimers, however, the resorcinarene provides a rigid scaffold and positions the OH groups in a way almost perfectly preorganized to form a complete seam of well-ordered hydrogen bonds. This overrides the unfavorable entropy encountered in water. Consequently, we suggest the HDX mechanism to be concerted in the case of the resorcinarene dimer.
Conclusions
The HDX data presented in this work allows us to draw the following conclusions:(a) There are significant exchange-rate differences depending on the cation size. Resorcinarene dimers which bear a fully closed seam of hydrogen bonds react much faster than those in which the seam is interrupted, e.g. the PacMan-shaped dimers or the heterodimer of non- and tetramethylated resorcinarene. The exchange-rate differences thus deliver structural insight: A fast exchange indicates an intact capsule with fully closed hydrogen-bonding seam.
(b) In order to explain all experimental results, a detailed consideration of the exchange mechanisms is required. HDX reactions can provide data on hydrogen bonding, but it would be too simplified to assume that non-hydrogen-bonded H atoms undergo readily an exchange, while those involved in hydrogen bonding do not.
(c) The concerted mechanistic scenario for the fast exchange on the Cs+-encapsulating resorcinarene and pyrogallarene dimers is an interesting one-dimensional Grotthuss mechanism. In particular, the question whether the preorganization of the OH groups through the resorcinarene and pyrogallarene scaffolds help to make the mechanism a concerted one might be of interest for studies with other methods complementary to mass spectrometry.
Acknowledgements
We thank Dr Andreas Springer for numerous fruitful discussions. Funding by the Deutsche Forschungsgemeinschaft (DFG), the German Academic Exchange Service (DAAD), the Fonds der Chemischen Industrie (FCI) and the Academy of Finland (KR, projects no. 212588 and 218325) is gratefully acknowledged. E.V.D. thanks the Studienstiftung des Deutschen Volkes for a PhD fellowship.Notes and reference
- C. A. Wraight, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 886–912 CrossRef CAS.
- J. E. Walker, Angew. Chem., Int. Ed., 1998, 37, 2308–2319 CrossRef.
- P. D. Boyer, Angew. Chem., Int. Ed., 1998, 37, 2296–2307 CrossRef.
- P. Agre, Angew. Chem., Int. Ed., 2004, 43, 4278–4290 CrossRef CAS.
- C. J. T. de Grotthuss, Ann. Chim., 1806, 58, 54–73 Search PubMed.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- T. J. F. Day, U. W. Schmitt and G. A. Voth, J. Am. Chem. Soc., 2000, 122, 12027–12028 CrossRef CAS.
- A. A. Kornyshev, A. M. Kuznetsov, E. Spohr and J. Ulstrup, J. Phys. Chem. B, 2003, 107, 3351–3366 CrossRef CAS.
- H. Lapid, N. Agmon, M. K. Petersen and G. A. Voth, J. Chem. Phys., 2005, 122, 014506–014511 CrossRef.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- I. Ohmine and S. Saito, Acc. Chem. Res., 1999, 32, 741–749 CrossRef CAS.
- Z. Luz and S. Meiboom, J. Am. Chem. Soc., 1964, 86, 4768–4769 CrossRef CAS.
- U. W. Schmitt and G. A. Voth, J. Chem. Phys., 1999, 111, 9361–9381 CrossRef CAS.
- J. B. Niederl and H. J. Vogel, J. Am. Chem. Soc., 1940, 62, 2512–2514 CrossRef CAS.
- V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
- H.-J. Schneider and U. Schneider, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 67–83 CrossRef CAS.
- L. C. Palmer and J. Rebek, Jr., Org. Biomol. Chem., 2004, 2, 3051–3059 RSC.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Frohlich, S. Kotila, C. Nather and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS.
- L. Avram and Y. Cohen, Org. Lett., 2006, 8, 219–222 CrossRef CAS.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 CrossRef CAS.
- W. Abraham, Journal of Inclusion Phenomena and Macrocyclic Chemistry, 2002, 43, 159–174 Search PubMed.
- N. K. Beyeh, M. Kogej, A. Åhman, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2006, 45, 5214–5218 CrossRef CAS.
- O. D. Fox, M. G. B. Drew, E. J. S. Wilkinson and P. D. Beer, Chem. Commun., 2000, 391–392 RSC.
- M. Nissinen, E. Wegelius, D. Falabu and K. Rissanen, CrystEngComm, 2000, 2, 151–153 RSC.
- Y. Aoyama, Y. Tanaka and S. Sugahara, J. Am. Chem. Soc., 1989, 111, 5397–5404 CrossRef CAS.
- Y. Aoyama, Y. Tanaka, H. Toi and H. Ogoshi, J. Am. Chem. Soc., 1988, 110, 634–635 CrossRef CAS.
- B. Botta, G. D. Monache, P. Picciardi, G. Zappia, C. Seri, E. Gacs-Baitz, P. Csokarsi and D. Misiti, Eur. J. Org. Chem., 2000, 10, 841–847 CrossRef.
- Y. Kikuchi, Y. Tanaka, S. Sutarto, K. Kobayashi, H. Toi and Y. Aoyama, J. Am. Chem. Soc., 1992, 114, 10302–10306 CrossRef CAS.
- K. Kobayashi, Y. Asakawa and Y. Aoyama, Supramol. Chem., 1993, 2, 133–135 CAS.
- K. Kobayashi, Y. Asakawa, Y. Kikuchi, H. Toi and Y. Aoyama, J. Am. Chem. Soc., 1993, 115, 2648–2654 CrossRef CAS.
- K. Kobayashi, M. Tominaga, Y. Asakawa and Y. Aoyama, Tetrahedron Lett., 1993, 34, 5121–5124 CrossRef CAS.
- H. Konishi and O. Morikawa, Chem. Express, 1991, 10, 801–804 Search PubMed.
- H. Mansikkamäki, M. Nissinen, C. A. Schalley and K. Rissanen, New J. Chem., 2003, 27, 88–97 RSC.
- H. Mansikkamaki, C. A. Schalley, M. Nissinen and K. Rissanen, New J. Chem., 2005, 29, 116–127 RSC.
- C. A. Schalley, Int. J. Mass Spectrom., 2000, 194, 11–39 Search PubMed.
- C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253–309 CrossRef CAS.
- B. Baytekin, H. T. Baytekin and C. A. Schalley, Org. Biomol. Chem., 2006, 4, 2825–2841 RSC.
- H. D. F. Winkler, E. V. Dzyuba and C. A. Schalley, New J. Chem., 2011 10.1039/c0nj00634c.
- H. A. Cox, R. R. Julian, S. W. Lee and J. L. Beauchamp, J. Am. Chem. Soc., 2004, 126, 6485–6490 CrossRef CAS.
- C. Lifshitz, Int. J. Mass Spectrom., 2004, 234, 63–70 Search PubMed.
- Z. Takats, S. C. Nanita, G. Schlosser, K. Vekey and R. G. Cooks, Anal. Chem., 2003, 75, 6147–6154 CrossRef CAS.
- U. Mazurek, O. Geller, C. Lifshitz, M. A. McFarland, A. G. Marshall and B. G. Reuben, J. Phys. Chem. A, 2005, 109, 2107–2112 CrossRef CAS.
- E. Kalenius, D. Moiani, E. Dalcanale and P. Vainiotalo, Chem. Commun., 2007, 3865–3867 RSC.
- E. Ventola, K. Rissanen and P. Vainiotalo, Chem.–Eur. J., 2004, 10, 6152–6162 CrossRef CAS.
- E. Kalenius, R. Neitola, M. Suman, E. Dalcanale and P. Vainiotalo, J. Am. Soc. Mass Spectrom., 2010, 21, 440–450 CrossRef CAS.
- S.-W. Lee, H.-N. Lee, H. S. Kim and J. L. Beauchamp, J. Am. Chem. Soc., 1998, 120, 5800–5805 CrossRef CAS.
- D. P. Weimann, H. D. F. Winkler, J. A. Falenski, B. Koksch and C. A. Schalley, Nat. Chem., 2009, 1, 573–577 CrossRef CAS.
- H. D. F. Winkler, D. P. Weimann, A. Springer and C. A. Schalley, Angew. Chem., Int. Ed., 2009, 48, 7246–7250 CrossRef CAS.
- T. Wyttenbach and M. T. Bowers, J. Am. Soc. Mass Spectrom., 1999, 10, 9–14 CrossRef CAS.
- S. Campbell, M. T. Rodgers, E. M. Marzluff and J. L. Beauchamp, J. Am. Chem. Soc., 1995, 117, 12840–12854 CrossRef CAS.
- J. I. Brauman, in Kinetics of Ion-Molecule Reactions, ed. P. Ausloos, Plenum Press, New York, 1979, pp. 153–164 Search PubMed.
- S. G. Lias, J. Phys. Chem., 1984, 88, 4401–4407 CrossRef CAS.
- E. Gard, M. K. Green, J. Bregar and C. B. Lebrilla, J. Am. Soc. Mass Spectrom., 1994, 5, 623–631 CrossRef CAS.
- P. Ausloos and S. G. Lias, J. Am. Chem. Soc., 1981, 103, 3641–3647 CrossRef CAS.
- X. Cheng and C. Fenselau, Int. J. Mass Spectrom. Ion Processes, 1992, 122, 109–119 CrossRef.
- M. Rozman, J. Am. Soc. Mass Spectrom., 2005, 16, 1846–1852 CrossRef CAS.
- M. Mäkinen, P. Vainiotalo and K. Rissanen, J. Am. Soc. Mass Spectrom., 2002, 13, 851–861 CrossRef CAS.
- D. M. Rudkevich, Chem.–Eur. J., 2000, 6, 2679–2686 CrossRef CAS.
- W. Saenger, C. Betzel, B. Hingerty and G. M. Brown, Nature, 1982, 296, 581–583 CrossRef CAS.
- S. W. Lee, H. N. Lee, H. S. Kim and J. L. Beauchamp, J. Am. Chem. Soc., 1998, 120, 5800–5805 CrossRef CAS.
- A. Åhman, M. Luostarinen, C. A. Schalley, M. Nissinen and K. Rissanen, Eur. J. Org. Chem., 2005, 13, 2793–2801 CrossRef.
- T. Gerkensmeier, C. Agena, W. Iwanek, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Z. Naturforsch. B, 2001, 56, 1063–1073 CAS.
- M. Luostarinen, A. Åhman, M. Nissinen and K. Rissanen, Supramol. Chem., 2004, 16, 505–512 CrossRef CAS.
- M. Luostarinen, K. Salorinne, H. Lähteenmäki, H. Mansikkamäki, C. A. Schalley, M. Nissinen and K. Rissanen, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 71–80 CrossRef CAS.
- S. A. Hofstadler, K. A. Sannes-Lowery and R. H. Griffey, J. Mass Spectrom., 2000, 35, 62–70 CrossRef CAS.
- M. C. Letzel, C. Agena and J. Mattay, J. Mass Spectrom., 2002, 37, 63–68 CrossRef CAS.
- A. Åhman, M. Luostarinen, K. Rissanen and M. Nissinen, New J. Chem., 2007, 31, 169–177 RSC.
- M. Mäkinen, J.-P. Jalkanen and P. Vainiotalo, Supramol. Chem., 2005, 17, 377–381 CrossRef.
- S. Mecozzi and J. Rebek, Jr., Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902–1903 RSC.
- S. J. Dalgarno, J. Antesberger, R. M. McKinlay and J. L. Atwood, Chem.–Eur. J., 2007, 13, 8248–8255 CrossRef.
- A. Shivanyuk, J. C. Friese, S. Döring and J. Rebek, Jr., J. Org. Chem., 2003, 68, 6489–6496 CrossRef.
- M. Fujio, R. T. McIver, Jr. and R. W. Taft, J. Am. Chem. Soc., 1981, 103, 4017–4029 CrossRef CAS.
- E. P. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef.
- P. J. Linstrom and W. G. Mallard (ed.), NIST webbook, National Institute of Standards and Technology, Gaithersburg MD, 20899, USA, http://webbook.nist.gov (retrieved Oct 21, 2010) Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |