Surface enhanced spatially offset Raman spectroscopic (SESORS) imaging – the next dimension
Nicholas
Stone
*a,
Marleen
Kerssens
a,
Gavin Rhys
Lloyd
a,
Karen
Faulds
b,
Duncan
Graham
b and
Pavel
Matousek
c
aBiophotonics Research Unit, Gloucestershire Hospitals NHS Foundation Trust, Great Western Road, Gloucester, GL1 3NN, UK. E-mail: n.stone@medical-research-centre.com; Fax: +44 8454 225485; Tel: +44 8454 225486
bCentre for Molecular Nanometrology, WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK
cCentral Laser Facility, Research Complex at Harwell, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0QX, UK
First published on 7th January 2011
Abstract
SESORS - Surface enhanced spatially offset Raman spectroscopy–imaging is explored for the first time in this study. Multiplexed surface enhanced Raman scattering (SERS) signals have been recovered non-invasively from a depth of 20 mm in tissues for the first time and reconstructed to produce a false colour image. Four unique ‘flavours’ of SERS nanoparticles (NPs) were injected into a 20 × 50 × 50 mm porcine tissue block at the corners of a 10 mm square. A transmission Raman data cube was acquired over an 11 × 11 pixel grid made up of 2 mm steps. The signals were reconstructed using the unique peak intensities of each of the nanoparticles. A false colour image of the relative signal levels was produced, demonstrating the capability of multiplexed imaging of SERS nanoparticles using deep Raman spectroscopy. A secondary but no less significant achievement was to demonstrate that Raman signals from SERS nanoparticles can be recovered non-invasively from samples of the order of 45–50 mm thick. This is a significant step forward in the ability to detect and identify vibrational fingerprints within tissue and offers the opportunity to adapt these particles and this approach into a clinical setting for disease diagnosis.
Introduction
Numerous advances in deep Raman techniques have led to the possibility of combining both surface enhanced Raman scattering (SERS) and deep Raman techniques together.1–3 The combination of SERS and spatially offset Raman spectroscopy (SORS) techniques, referred to as SESORS, opens the way for sampling a number of disease conditions in the same organ at the same time, thus potentially leading to a new methodology for enhanced personalised treatment plans to be developed in real-time. Here we explore the possibility of using this method to provide multiplexed imaging for the first time at depths of greater than 20 mm in tissues.SERS can provide molecularly specific enhancement of Raman signals,4,5 when the target molecule and a roughened (nanometre scale) noble metal surface are brought in close proximity to one another. Enhancement factors on the order of 109 are possible and single molecule detection has been reported.6,7 A lack of reproducibility of signals has limited uptake in a clinical environment. However, recent developments of novel substrates, such as encapsulated nanoparticles8 and photonic crystals9,10 show great promise in overcoming these difficulties.
Further developments have included the use of surface enhanced resonance Raman scattering (SERRS), a resonance SERS technique pioneered by Stacy and Van Duyne11 and developed for use as a clinical tool by Graham et al.,12 which is able to provide equivalent detection limits to fluorescence labelled dyes.13 It has become routine to tag SERS nanoparticles (NPs) with antibodies for molecules of interest and numerous groups are exploring the clinical application of this approach. In the cancer environment, tagged nanoparticles enhancing specific signals from malignant markers, are either being used in vivo14–16 or as molecular specific stains for histopathology;17–19 with the possibility of numerous multiplexed SERS/SERRS stains providing hyperspectral images of the locations of molecules of interest from the same spectral acquisition and tissue slice.20
Until very recently it was only considered possible to measure signals from these NPs at maximum depths of around 5 mm.15 The majority of the work performed to date has explored SERS use ex vivo with the recent advances into in vivo animal based studies. However, these have generally been measuring SERS nanoparticles injected or accumulating at the surface of the animal.14–16
An in vivo study demonstrating the use of multiplexed tags within a mouse model was able to demonstrate the detection of 1.6 × 107nanoparticles subcutaneously and 3.1 × 107nanoparticles following intravenous injection.15 Furthermore this study demonstrated the maximum penetration depth achieved to date with SERS and traditional Raman microscopy was of the order of 5.5 mm with a bolus of 1.6 × 1011nanoparticles.
Rather than rely upon confocal techniques able to probe depths of up to around 200 μm in turbid media with conventional non-SERS samples, deep Raman techniques have been able to distinguish chemical composition of samples through around 2.5 cm thick or 100 times deeper.21 The limits have theoretically been extrapolated to 4–5 cm of tissue thickness with proposed optimisation of instrumentation.22 This rapidly expanding field is leading towards many novel potential applications for in vivo medical diagnostics from probing bone composition through the skin to disease specific breast calcifications and soft tissue lesions for cancer diagnostics.23–27 A further possibility includes in vivo monitoring of drug delivery and identification of the location of the drug interactions.
Our recent paper introducing the concept of SESORS, reached a key milestone in deep Raman spectroscopy by demonstrating the possibility of probing signals from SERS nanoparticles buried, or injected into tissues from depths significantly deeper (25 mm) than that previously achieved in epi-Raman approaches.28 In this work we used transmission Raman spectroscopy to demonstrate the concept of deep Raman spectroscopy. This method can be considered a special form of spatially offset Raman spectroscopy (SORS) where the collection and laser delivery areas are displaced to the extreme, i.e. being on the opposite sides of the sample. In this study it was possible to detect 1.1 × 109 NPs through 25 mm of porcine tissue. This was achieved at a depth of around five times that achieved by Keren et al.15 and around 100 times less SERS NPs.
A recent paper by Van Duyne's group29 expands on the proposed SESORS approach by exploring the potential of sensing glucose concentration subcutaneously in tissue fluid. They use a similar configuration to the one that we have described for signal collection coupled to an implanted silver film over a nanosphere substrate. They found that it was possible to achieve mean absolute relative difference values for calibration of 16.6% and validation 34.6%. Higher error RMSEC of 58.11 mg dL−1 (3.23 mM) and RMSEP of 96.35 mg dL−1 (5.35 mM) was seen than in previous in vivo results by the same group. This is not unexpected considering the added optical dispersion of the skin and the displacement of the sensor during the experiment.
In this paper, we take a significant step by adding not only the capability to probe nanoparticles at depth in tissues, but to image multiple ‘flavours’ in the same sample. This demonstrates the potential for multiplexed sensing capability to distinguish both spatially and spectrally between several different nanoparticles.
The postulated depth limit for recovery of signals with transmission Raman (as outlined in previous publications) was around 4–5 cm.22 An additional experiment described here has explored the maximum depth in tissues through which a recognisable signal from the SERS NPs could be obtained at a depth of around 5 cm.
Encapsulated SERS active nanoparticles produced by Cabot (formerly Oxonica) (Boston, Massachusetts) were used in this study.8 In contrast to our previous study, these SERS active nanoparticles consisted of gold particles coated with Raman reporters which were optimised for use with 785 or 830 nm excitation and were encapsulated in a silica shell. One of the benefits of the use of encapsulated SERS nanoparticles is that the signals are obtained only from the reporter molecule, rather than for the target molecule of interest. This allows us to demonstrate the performance of the technology using NPs which would produce the same signals when conjugated directly with molecules of diagnostic interest. They could be labelled for numerous targets, such as PSA, p53 proteins, DNA-fragments and cell specific proteins. This reveals the prospects for numerous clinical applications such as early cancer detection and staging, metastatic detection, treatment monitoring and chemo-sensitivity, provided that the disease specific target molecule is known.30
Results and discussions
Nanoparticle signals from four distinct flavours were identified using the peak intensities unique (within this set) to each of the four nanoparticles flavours. These were 1385, 1300, 928, and 1645 cm−1 for the Cabot NPs flavours 403, 421, 422, 440 respectively (see experimental section for SERS reporter identities). Fig. 1 shows spectra measured from the NP suspensions air dried and measured at 830 nm on a calcium fluoride slide.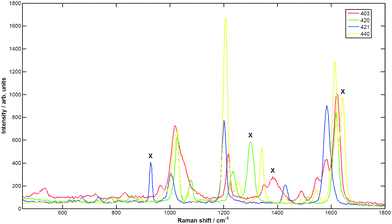 | ||
| Fig. 1 Spectra at 830 nm of Cabot NPs. Red is x403, green is x420, blue is x421, yellow is x440. The Xs signify the identity peaks used to measure the NP flavour signal in the SESORS spectrum. | ||
Injecting the four flavours into a large porcine tissue block (20 × 50 × 50 mm) enabled the spatial distribution of the spectral signals to be measured and visualised. The signal level for each pixel using the distinct NP identity peaks relative to the quiet spectral region intensity at 800 cm−1 was calculated. This produced a four-layered matrix, where each layer corresponded to the relative signal from each NP flavour. False colour map images of the relative signal levels were constructed. Fig. 2 shows the distribution of the signals from each individual nanoparticle flavour (right) and a combined image with all flavours displayed together. This clearly demonstrates the capability of multiplexed imaging of SERS nanoparticles using SESORS.
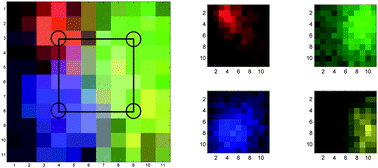 | ||
| Fig. 2 False colour images of the SERS NP signals, measured in a 11 × 11 grid, pixel size 2 mm. Left image shows all signals plotted together (with pixel colour mix showing combined signals) with the approximate injection points marked, right image shows each ‘flavour’ separately. Red is x403, green is x420, blue is x421, yellow is x440. | ||
The images shown were reconstructed from peak intensities of the most distinct SERS bands from each NP flavour. Other multivariate approaches were also explored, such as principal component analysis and least squares fitting of the pure spectra to the SESORS spectra, but produced similar results (not shown). If more flavours of NPs were used, it is expected that these approaches may become more important.
It should be noted that the maximum injected volume was 13 μL for each flavour which is equivalent to around a 2 mm diameter droplet, the same as a single pixel in the image. Each droplet contained a maximum of 1.8 × 109 NPs. It is clear from the images in Fig. 2, that the diffuse scattering of the Raman photons produced by the NPs has the effect of increasing the apparent spot sizes, i.e. the spot areas that the signals are collected from at the surface of the tissue, when measured through the tissue. The lateral spatial resolution in transmission Raman spectroscopy deteriorates linearly with sample thickness and could be typically expected to be approximately equal to a half of the sample thickness.31,32 To investigate this a Gaussian peak shape was fitted to each of the NP layers used to generate the pseudo-colour image. This was achieved using a simplex search method to minimise the residual error between the fitted peak and the raw data using five peak parameters: the intensity of the maximum, the position of the maximum (both x and y coordinates), the width (σ) and an offset. The full width at half maximum (FWHM) was then calculated from σ as follows:  using a standard Gaussian function.33 The FWHM for NPs 403, 421, 422, 440 were calculated to be 7.6, 13.6, 12 and 18 mm respectively. Although these values are subject to various sources of error, such as assuming that a Gaussian is a suitable shape to fit the data, they are within the expected range for this experiment. Other potential errors may come from accidental variation of the depth of injection and diffusion of the NPs across natural voids in heterogeneous tissue. Further to the deterioration in localisation of the source scatterers with sample thickness, there is significant signal mixing in deeper tissues due to the high turbidity of tissues at NIR wavelengths. However, by using the approach outlined it was a trivial process to separate the spectral signals of the SERS NPs.
using a standard Gaussian function.33 The FWHM for NPs 403, 421, 422, 440 were calculated to be 7.6, 13.6, 12 and 18 mm respectively. Although these values are subject to various sources of error, such as assuming that a Gaussian is a suitable shape to fit the data, they are within the expected range for this experiment. Other potential errors may come from accidental variation of the depth of injection and diffusion of the NPs across natural voids in heterogeneous tissue. Further to the deterioration in localisation of the source scatterers with sample thickness, there is significant signal mixing in deeper tissues due to the high turbidity of tissues at NIR wavelengths. However, by using the approach outlined it was a trivial process to separate the spectral signals of the SERS NPs.
The particle numbers used in both this and our previous SESORS study28 were very similar, although the signal to noise achieved with these NPs was significantly greater. Raman spectra measured in transmission mode for particles injected into around 20 mm and 47 (45–50) mm thick blocks are shown in Fig. 3. It can be seen from the raw spectra that SERS signals can be identified clearly at both thicknesses, although the 47 mm specimen required more particles (around 20 times more) and a longer collection time (5 × 60 s) to achieve this effect. Signals recovered show an order of magnitude increase in depth with an order of magnitude less NPs than shown in previous state-of-the-art results.15
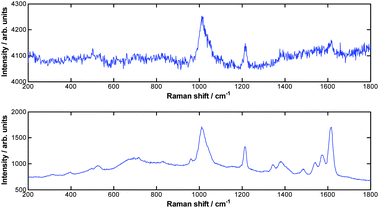 | ||
| Fig. 3 Raw spectra of NP flavour x403 with 3 × 1010 particles in 50 μL for 47 mm (range 45–50 mm) × 50 × 50 mm tissue block (top frame) and 1.8 × 109 particles in 3 μL for 20 mm thick × 50 × 50 mm tissue block (bottom frame). | ||
One feature of the results to note is that the SESORS spectra measured through 47 mm of tissue show significant signal loss, particularly above 1250 cm−1, which we had not previously observed with tissue. We propose the following mechanism; at these wavelengths both water and myoglobin contribute to the absorption. The 1250 to 1600 cm−1 Stokes-shifted range with an 830 nm excitation equates to around 925 to 957 nm, which covers the range where a strong lipid absorption band (∼930nm)34 and water absorption band (max ∼970nm) are located.35 In addition a more subtle contribution also comes from a rising absorption in this wavelength region from myoglobin.36
Experimental
Apparatus
The deep Raman system utilised for this study was constructed at Gloucestershire Royal Hospital using the same configuration as outlined in previous papers.27,22 This includes an Innovative Photonics Solutions 830 nm laser as the source which was filtered with two 830 nm laser line filters (Thorlabs), leaving a collimated spot of 4 mm diameter and 219 mW at the sample surface.The transmitted Raman light was collected using a standard 50 mm diameter fused silica lens with a focal length of 60 mm. The scattered light was collimated and passed through a 50 mm diameter holographic notch filter (830 nm, Kaiser Optical Systems, Inc.) to suppress the elastically scattered component of light. The second lens, identical to the first, was then used to image, with magnification 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the sample interaction zone onto the front face of a fibre bundle. The laser incident spot was positioned in such a way so that it coincided with the centre of the fibre probe axis projected through the imaging system on the sample.
:1, the sample interaction zone onto the front face of a fibre bundle. The laser incident spot was positioned in such a way so that it coincided with the centre of the fibre probe axis projected through the imaging system on the sample.
The fibre bundle collecting the Raman light consisted of 22 active fibres made of silica with a core diameter of 220 μm, a doped silica cladding diameter of 240 μm and a polyimide coating of 265 μm diameter. The fibre numerical aperture was 0.37. The bundle was custom made by CeramOptec Industries, Inc. The Raman scattered light was propagated through the fibre bundle of length ∼2 m to the end with the fibres mounted in a linear array, which was oriented vertically and placed in the input image plane of a Kaiser Optical Technologies Holospec 1.8i NIR spectrograph. Raman spectra were collected using a NIR back-illuminated deep-depletion TE cooled (−80 °C) CCD camera (Andor Technology, DU420A-BR-DD, 1024 × 256 pixels) by binning the entire chip vertically.
Samples
Tissue preparation involved collection of fresh porcine samples from the local abattoir and dissecting them to the required dimensions. Muscle tissue was chosen to represent dense human tissues as discussed elsewhere.22 A demonstration of signal obtained through increasing depth was achieved by cutting samples from pork muscle to thicknesses of around 20 and 50 mm (other dimensions 50 × 50 mm) and mounted in the experimental apparatus. All dimensions were measured approximately using a basic ruler at a number of points and mean thicknesses were calculated.SESORS multiplexing
Four flavours of NPs (Cabot x403 (5-(4-pyridyl)-1,3,4-oxadiazole-2-thiol - POT), x420 (4,4′-dipyridyl - DIPY), x421 (d8-4,4′-dipyridyl - d8DIPY) and x440 (Trans-1,2-Bis(4-pyridyl)-ethylene - BPE) were used in this multiplexing experiment. Their construction is described in detail elsewhere;8 briefly, they include a gold core of around 100 nm diameter surrounded by a reporter molecule, the whole encapsulated in a thin silica layer. The particles were delivered suspended in water.The suspension was shaken for one minute prior to pipetting the required volume into a syringe for injection into the tissue. Both 3 μL of NP stock solution plus 10 μL pestanal water (Sigma) (1.8 × 109 NPs) were pipetted into a 10 mL syringe with a 21G needle of inner diameter 0.51 mm (other needles of finer bore were initially tested and they were found to prevent the majority of the NPs from passing through). Each NP suspension was injected into points at the corners of a 10 mm square, to approximately 50% of the depth of the tissue. It was not possible to be certain that some NPs did not remain in the syringe so the number of NPs pipetted into the syringe prior to injection should be taken as a maximum value. Fig. 4 shows the template grid used. The tissue block (and the NP grid square) was then centered in the sample compartment. The template was then removed, prior to taking SESORS measurements.
 | ||
| Fig. 4 Left: 20 × 50 × 50 mm porcine block with grid used to align injection points for NPs. The image grid was acquired over the 20 × 20 mm square shown by the template (removed for measurements). Right tissue mounting, laser fibre (400 μm core), 2 laser line filters and the laser spot and scattering on the surface of the tissue mounted in the hollow anodised metal bracket. | ||
Translational x-y stages with a range of 20 mm were used to translate the sample in steps of 2 mm to construct an image of 11 × 11 pixels. Time of collection was 10 s at each point. Fig. 4 also shows the tissue mounting, laser fibre (400 μm core), 2 laser line filters and the laser spot and laser back-scattering at the surface of the tissue, which was mounted in a hollow anodised metal bracket.
Extended depth SESORS measurements
For this experiment a single SERS NP flavour (x403) was selected and a large number of particles (3 × 1010) were injected into the approximate centre of a porcine muscle tissue block of 45–50 mm (mean 47 mm) thick by 50 × 50 mm. Fig. 5 shows the side projection of the 47 mm tissue block in the experimental position, the illumination provided on one side and the collection at the other side of the tissue block. The signal was measured in 5 × 60 s and compared with that achieved using the imaging block (20 mm thick) with 1.8 × 109 particles. | ||
| Fig. 5 47(45–50) × 50 × 50 mm tissue block used for the extended depth SESORS experiment. | ||
Conclusions
Multiplexed SERS signals have been recovered from 20 mm in tissues for the first time and reconstructed to produce a false colour image. The signals were reconstructed using the unique peak intensities of each of the nanoparticle flavours. A false colour map image of the relative signal levels was constructed, demonstrating the capability of multiplexed SESORS imaging of SERS nanoparticles using transmission Raman spectroscopy. A secondary but no less significant achievement was to demonstrate that Raman signals from SERS nanoparticles can be recovered from samples of the order of 50 mm thick. This is double that achieved in the first demonstration of this method (although with around 20–30 times as many particles).The prospects for SESORS as a medical tool are significant. There are numerous applications where this approach would have a major impact on rapid specific diagnosis, patient specific treatment selection and treatment monitoring. However, in addition to further developments in disease specific SERS particles and read out methodologies; the greatest hurdle will be the issue of introducing nanoparticles into the body without yet a full understanding of their excretion mechanism or long term accumulation sites and whether this is likely to have any detrimental effects.
Acknowledgements
The authors would like to acknowledge the support of Michael Natan and Cabot in providing the NPs used in this study.Nick Stone holds a Senior Research Fellowship (Career Scientist) from the National Institute of Health Research. Marleen Kerssens is jointly funded by Gloucestershire Hospitals NHS Foundation Trust and the Science and Technology Facilities Council Biomedical Network (4161234). Duncan Graham and Karen Faulds acknowledge the awards of a Science and Innovation and Platform Grant from the EPSRC to support this work.
References
- P. Matousek, I. P. Clark, E. R. C. Draper, M. D. Morris, A. E. Goodship, N. Everall, M. Towrie, W. F. Finney and A. W. Parker, Appl. Spectrosc., 2005, 59, 393–400 CrossRef CAS.
- P. Matousek, M. D. Morris, N. Everall, I. P. Clark, M. Towrie, E. Draper, A. Goodship and A. W. Parker, Appl. Spectrosc., 2005, 59, 1485–1492 CrossRef CAS.
- N. A. Macleod and P. Matousek, Appl. Spectrosc., 2008, 62, 291A–304A CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef CAS.
- K. Kneipp et al., Phys. Rev. Lett., 1997, 78, 1667 CrossRef CAS.
- J. A. Dieringer, K. L. Wustholz, D. J. Masiello, J. P. Camden, S. L. Kleinman, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2009, 131, 849–854 CrossRef CAS.
- W. E. Doering, M. E. Piotti, M. J. Natan and R. G. Freeman, Adv. Mater., 2007, 19, 3100–3108 CrossRef CAS.
- S. Cintra et al., Faraday Discuss., 2006, 132, 191–199 RSC.
- S. Mahajan et al., Phys. Chem. Chem. Phys., 2007, 9, 104–109 RSC.
- A. A. Stacy and R. P. Van Duyne, Chem. Phys. Lett., 1983, 102, 365–370 CrossRef CAS.
- D. Graham, W. E. Smith, A. M. T. Linacre, C. H. Munro, N. D. Watson and P. C. White, Anal. Chem., 1997, 69, 4703–4707 CrossRef CAS.
- G. Sabatte, R. Keir, M. Lawlor, M. Black, D. Graham and W. E. Smith, Anal. Chem., 2008, 80, 2351–2356 CrossRef CAS.
- A. Pal, N. R. Isola, J. P. Alarie, D. L. Stokes and T. Vo-Dinh, Faraday Discuss., 2006, 132, 293–301 RSC.
- S. Keren, C. Zavaleta, Z. Cheng, A. de la Zerda, O. Gheysens and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5844–5849 CrossRef CAS.
- X. M. Qian et al., Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS.
- L. Sun et al., Nano Lett., 2007, 7, 351–356 CrossRef CAS.
- B. Lutz et al., J. Histochem. Cytochem., 2008, 56, 371–379 CAS.
- Y. N. Liu, Z. O. Zou, Y. O. Liu, X. X. Xu, G. Yu and C. Z. Zhang, Spectroscopy and Spectral Analysis, 2007, 27, 2045–2048 Search PubMed.
- K. Faulds, R. Jarvis, W. E. Smith, D. Graham and R. Goodacre, Analyst, 2008, 133, 1505–1512 RSC.
- P. Matousek and N. Stone, Analyst, 2009, 134, 1058 RSC.
- N. Stone and P. Matousek, Cancer Res., 2008, 68, 4424–4430 CrossRef CAS.
- M. V. Schulmerich, Proceedings of SPIE, 2006, 6093, 60930O Search PubMed.
- M. V. Schulmerich, K. A. Dooley, M. D. Morris, T. M. Vanasse and S. A. Goldstein, J. Biomed. Opt., 2006, 11, 060502 CrossRef.
- P. Matousek et al., Appl. Spectrosc., 2006, 60, 758 CrossRef CAS.
- M. V. Schulmerich, J. Biomed. Opt., 2008, 13, 020506 CrossRef.
- P. Matousek and N. Stone, J. Biomed. Opt., 2007, 12, 024008 CrossRef.
- N. Stone, K. Faulds, D. Graham and P. Matousek, Anal. Chem., 2010, 82, 3969–3973 CrossRef CAS.
- J. M. Yuen, N. C. Shah, J. T. Walsh, Jr., M. R. Glucksberg and R. P. Van Duyne, Anal. Chem., 2010, 82, 8382–8385 CrossRef CAS.
- C. G. Rao et al., Int. J. Oncol., 2005, 27, 49–57.
- N. Everall, P. Matousek, N. Macleod, K. L. Ronayne and I. P. Clark, Appl. Spectrosc., 2010, 64, 52–60 CrossRef CAS.
- N. Everall, I. Priestnall, P. Dallin, J. Andrews, I. Lewis, K. Davis, H. Owen and M. W. George, Appl. Spectrosc., 2010, 64, 476–484 CrossRef CAS.
- Eric W. Weisstein, “Gaussian Function”. From MathWorld–A Wolfram Web Resource. http://mathworld.wolfram.com/GaussianFunction.html.
- S. Kukreti, A. Cerussi, B. Tromberg and E. Gratton, Dis Markers., 2008, 25, 281–290 Search PubMed.
- S. J. Matcher, M. Cope and D. T. Delpy, Phys. Med. Biol., 1994, 39, 177 CrossRef CAS.
- J. J. Xia, E. P. Berg, J. W. Lee and Yao, Meat Sci., 2007, 75, 78–83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |