DNA-programmed spatial screening of carbohydrate–lectin interactions†
Christian
Scheibe
a,
Alexander
Bujotzek
b,
Jens
Dernedde
c,
Marcus
Weber
b and
Oliver
Seitz
*a
aInstitut für Chemie der Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489, Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de; Fax: (+49) 30 2093 7266
bZuse Institute Berlin, Takustraße 7, 14195, Berlin, Germany
cZentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité-Universitätsmedizin Berlin (CBF), Hindenburgdamm 30, 12203, Berlin, Germany
First published on 25th January 2011
Abstract
A wide range of multivalent scaffolds was assembled by using only five different PNA oligomers and various DNA templates. The flexibility of the PNA–DNA duplexes could be increased by introducing nick-sites and partially unpaired regions, as confirmed by MD simulations. The self-organized glyco-assemblies were used in a spatial screening of accessible carbohydrate binding sites in the Erythrina cristagalli lectin (ECL). This systematic investigation revealed a distance dependence which is in agreement with the crystal structure analysis.
Introduction
The interactions between proteins and carbohydrate ligands are often characterized by low binding affinities. In a cellular context, multivalent presentation of both carbohydrate ligands and carbohydrate binding sites compensate for weak interaction of an individual receptor–ligand pair.1,2 The binding of multivalent inhibitors is not only governed by the number but also by the distance and orientation between the presented carbohydrate ligands. Significant efforts have been invested in the synthesis of low-valent glyco-structures to precisely control the presentation of the individual glyco-ligands and to interrogate the spatial arrangement of the binding sites of the lectin.3 Highest binding affinities are usually achieved by using high-valent glyco-clusters based on multivalent scaffolds such as polymers,4dendrimers,5 and nanoparticles.6 A precise control of number, orientation, and distance between the presented carbohydrates is often difficult to realize in these glyco-clusters.Nucleic acid architectures offer intriguing opportunities for the multivalent presentation of glyco-ligands because (a) oligonucleotide synthesis provides monodisperse material, (b) the valency of the ligand display can readily be controlled by programmed self-assembly based on nucleic acid hybridization and (c) the well-known base-pairing rules allow Ångström-scale positioning of functional groups along the rigid nucleic acid helix. Kobayashi explored self-organized, high molecular weight DNA-galactose clusters of high periodicity and studied the effect of the helical torsion of glycan display on cooperative lectin recognition.7 Very recently, Winssinger described the hybridization of end-labeled peptide nucleic acids (PNA) carrying flexibly tethered bivalent hexamannose units.8 This study suggested that nucleic acid hybridization can be used to emulate complex carbohydrate epitopes. However, the ability to use DNA-programmed multivalency as a tool for the interrogation of the spatial arrangement of lectin binding sites has not been demonstrated yet.
Many lectins arrange binding sites over a convex surface calling for ligand displays from flexible or concave scaffolds.
Previous systematic screenings of distance dependencies in multivalent systems have relied on flexible scaffolds,9–11cyclodextrins,12 and cyclopeptides.13,14 Here we report the first example in which self-organized, supramolecular nucleic acid structures are used to scan the spatial arrangement of carbohydrate binding sites. The nucleic acid architectures were designed to contain semi-rigid regions in order to avoid torsional and bending stress upon multivalent recognition by convex lectin surfaces. The approach relies on PNA conjugates I which contain O-linked N-acetyl-lactosamine (LacNAc) residues at internal positions (Fig. 1). The DNA-instructed formation of multivalent complexes such as II or III was used to probe the distance of binding sites in the LacNAc-specific Erythrina cristagalli lectin (ECL).15
 | ||
| Fig. 1 Modular assembly of multipartite PNA–DNA complexes to control the valency and spatial arrangement of a multivalent display of glyco-ligands (yellow). | ||
Results and discussion
We have used peptide nucleic acids (PNA)16 as scaffold because PNA–DNA duplexes have a higher stability than DNA–DNA duplexes. This helps to maintain the integrity of the glyco-assemblies at low concentration. Furthermore, PNA is not susceptible to enzymatic degradation by nucleases, which would facilitate studies in biological environments.17 The construction of the multivalent LacNAc assemblies reported here involves automated PNA synthesis (Scheme 1), the modified cysteine-derived PNA monomer 118 which allows conjugation of LacNAc residues via thioether formation (Scheme 2), and hybridization of the obtained PNA oligomers to complementary DNA strands. The modularity of the approach depicted in Fig. 1 enables the rapid and facile assembly of a wide range of supramolecular structures with defined multivalency. The distances between the attached LacNAc ligands were estimated based on the known NMR structure of PNA–DNA duplexes, i.e. a helical twist that comprises 13 base pairs with a pitch of 42 Å.19 Accordingly, PNA oligomers of 13 base pairs length were synthesized. Nevertheless, the maleimido-containing linker and the nick sites in complexes such as III were expected to provide flexibility for the partial compensation of discrepancies in parallel presentation of LacNAc ligands arising from helical torsion.![Synthesis of thiol-modified PNA 5 and 6.
Reagents and conditions: (a) piperidine/DMF; (b) Fmoc-BBhoc-OH, HCTU, NMM; (c)
Ac2O/2,6-lutidine/DMF; (d) piperidine/DMF; (e)
TFA/EDT/iPr3SiH/H2O (Bhoc, benzhydryloxycarbonyl; EDT, ethanedithiol; HCTU, 5-chloro-1-[bis(dimethylamino)methylene]-1H-benzotriazolium-3-oxide
hexafluorophosphate; NMM, N-methylmorpholine).](/image/article/2011/SC/c0sc00565g/c0sc00565g-s1.gif) | ||
| Scheme 1 Synthesis of thiol-modified PNA 5 and 6. Reagents and conditions: (a) piperidine/DMF; (b) Fmoc-BBhoc-OH, HCTU, NMM; (c) Ac2O/2,6-lutidine/DMF; (d) piperidine/DMF; (e) TFA/EDT/iPr3SiH/H2O (Bhoc, benzhydryloxycarbonyl; EDT, ethanedithiol; HCTU, 5-chloro-1-[bis(dimethylamino)methylene]-1H-benzotriazolium-3-oxide hexafluorophosphate; NMM, N-methylmorpholine). | ||
 | ||
| Scheme 2 Synthesis of PNA-LacNAc conjugates. Reagents and conditions: (a) N-Cbz-2-aminoethanol, dimethyl(thiomethyl)sulfonium triflate, CH2Cl2, 4 Å MS; (b) Zn, HOAc; (c) Ac2O, pyridine; (d) cat. NaOMe, MeOH; (e) UDP-Gal, β1,4-galactosyltransferase, calf intestine alkaline phosphatase, bovine serum albumin, MnCl2, 0.1 M HEPES pH 7.0, 37 °C, 2 d; (f) H2, Pd/C; (g) N-succinimidyl-3-maleimidopropionate, NaHCO3, H2O/dioxane; (h) 5 or 6, 10 mM NaH2PO4, pH 6.5, 3 mM P(CH2CH2COOH)3. | ||
Automated solid-phase synthesis provided the thiol-modified PNA oligomers 5 and 6, as well as unmodified PNA oligomers 7, 8 and 9 (Scheme 1). The sequences of PNAs 5 and 7 were identical and so were 6 and 8. PNA 9 spanned a third sequence. The three different PNA-sequences were envisioned to serve as three independent anticodons for the recognition of codon segments on the DNA-template. The synthesis of the LacNAc building block was commenced from the thioglycosyl donor 10, which was used in the glycosylation of Cbz-protected aminoethanol (Scheme 2). Removal of the Troc-group, followed by acetylation of the liberated amine and subsequent deprotection of the hydroxyl groups afforded glycosyl acceptor 12. The N-acetyl-lactosamine 13 was established in a chemoenzymatic reaction which involved UDP-galactose and β1,4-galactosyltransferase. Advantages of this approach are high stereo- and regioselectivities, avoidance of laborious protection/deprotection steps, and high yields. After hydrogenolytic cleavage of the Cbz-group, the maleimido group was introduced. The conjugation of the LacNAc-maleimido building block 14 to thiolated PNA oligomers 5 and 6 was performed in the presence of triscarboxyethylphosphane to avoid disulfide formation.
The five different PNA-oligomers offered three independent anticodon sequences. DNA-templates that provide three codon segments distributed over four positions were used for the instructed formation of PNA–DNA complexes. In principle, 324 complexes can be formed by such a set-up. However, only a fraction was explored and the affinity for Erythrina cristagalli lectin (ECL) was tested by surface plasmon resonance (SPR). ECL was chosen as a multivalent model substrate because its carbohydrate binding affinities and structure have been well characterized.15 This lectin exists as a dimer in which two monomers adopt a handshake motif by associating back-to-back. Each monomer has one binding site for galactose-containing carbohydrates, including N-acetyl-lactosamine. The crystal structure revealed a 65 Å distance between the two binding sites. However, the binding sites are located on opposite sides of the protein. A multivalent substrate has to bend around the convex surface of the protein and the real distance required for simultaneous binding is larger, i.e. approximately 100 Å.
For initial SPR experiments the density of immobilized lectin on the gold surface was set to approximately 2700 RU (1 RU corresponds to ca. 1 pg lectin per mm2)20 and the influence of the number of LacNAc ligands on the binding behavior was investigated. As expected, no binding was detected for the PNA–DNA duplex carrying no LacNAc ligands (Fig. S2, ESI†). The PNA–DNA complex 17 was assembled through hybridization of a 52mer DNA with three different PNA oligomers (7, 9 and 16). The formed complex presented only a single LacNAc residue. The SPR sensorgram revealed the expected low affinity (KD = 800 μM) for the lectin (Table 1, Fig. 2). The complex 18 was assembled from two different PNA–LacNAc conjugates (15 and 16) and two identical unlabeled PNA oligomers 9. The SPR measurements suggested a 33-fold enhancement of binding affinity per LacNAc ligand. In the progression from mono- to di-, tri- and tetravalent presentation, the relative binding affinity per LacNAc ligand increased (Table 1, Fig. 2). Impressively, the LacNAc ligands of the tetravalent complex 20 (KD = 1.1 μM) have a 182-fold greater affinity than in the monovalent complex 17. Interestingly, the polyvalent cluster 21 formed by hybridization of sticky ends showed dramatically reduced dissociation rates; a behavior known from high-valent glyco-clusters21 (compare with sensorgram of asialofetuin, Fig. S1, ESI†).
| Complexa | k a/M−1s−1 | k d/s−1 | K D/μM | Relative potency | |
|---|---|---|---|---|---|
a
 = 7 or 15, = 7 or 15,  = 8 or 16, = 8 or 16,  = 9, = 9,  = complementary DNA, = complementary DNA,  =
LacNAc.
b exact valency not determined (ka,
association rate constant; kd, dissociation rate constant;
KD, dissociation constant. Conditions: flow =
20 μL min−1, 90 s association, 240 s dissociation.
Template sequences are detailed in ESI1). =
LacNAc.
b exact valency not determined (ka,
association rate constant; kd, dissociation rate constant;
KD, dissociation constant. Conditions: flow =
20 μL min−1, 90 s association, 240 s dissociation.
Template sequences are detailed in ESI1).
|
|||||
| 17 |

|
4.0 × 102 | 3.2 × 10−1 | 800 | 1 |
| 18 |

|
4.1 × 103 | 5.0 × 10−2 | 12 | 33 |
| 19 |

|
6.0 × 103 | 1.5 × 10−2 | 2.6 | 102 |
| 20 |

|
6.8 × 103 | 7.7 × 10−3 | 1.1 | 182 |
| 21 |

|
9.4 × 102 | 2.3 × 10−3 | 2.5 | —b |
 | ||
| Fig. 2 SPR sensorgrams showing the interactions between different LacNAc-PNA–DNA complexes (see Table 1) and surface-bound ECL (conditions: c = 10 μM, flow = 20 μL min−1, 90 s association, 240 s dissociation). | ||
We decided to investigate the binding behavior of the bivalent substrates in more detail. In the complexes 18 and 22–27, the distance between the LacNAc ligands was varied between approximately 42 Å in 22 and 146 Å in 27 (Fig. 3). Initially, only little to no differences between the binding affinities were observed (Fig. 3, grey bars). We assumed that the binding of the bivalent substrates not only involved the simultaneous recognition of the two binding sites on a single ECL molecule but also the interaction across two adjacent ECL molecules. The latter leads to cross-linking of lectin molecules, often referred to as aggregation, and can govern binding arrangements at high concentration.22 With the intention of reducing cross-site binding, the density of immobilized ECL was reduced to 700 RU. Indeed, the low loaded sensor chip uncovered the differences among the binding behavior of bivalent assemblies 18 and 22–27. The dissociation constant decreased as the LacNAc–LacNAc distance was increased from 42 to 104 Å and reached a minimum before further increases in distance to 146 Å resulted in increased dissociation constants (Fig. 3, black bars). The minimum can be attributed to a LacNAc spacing that is close to the spatial arrangement (ca. 100 Å) of the two binding sites of ECL.
 | ||
| Fig. 3 Influence of the LacNAc–LacNAc distance on the binding affinities of bivalent complexes 18 and 22–27 (see Table 1 for complex composition and ESI† for kinetic parameters). | ||
In addition to the distance between the LacNAc ligands, the flexibility of the complexes is expected to influence binding to ECL. It was envisioned that the three nick sites introduce local flexibility which is required to facilitate bending of the PNA–DNA complex around the protein. The complexes 24 and 25 were included in the study to investigate a further decrease of substrate rigidity since the unpaired regions of the DNA template are more flexible than paired regions. In fact, these substrates (24 and 25) showed the smallest dissociation constants of all tested bivalent substrates. Unpaired single strand regions (in 24 and 25) should bend more readily than nick sites in completely base-paired complexes (in 18, 22, 23, 26 and 27). It is thus plausible to assume that the flexible assemblies 24 and 25 better accommodate the curved shape required for simultaneous binding to ECL's carbohydrate binding sites. We concluded that substrates with LacNAc distances too short for simultaneous recognition by a single lectin molecule are limited to cross-site binding, which is less prevalent at reduced ECL surface loading. Assemblies exceeding the optimal LacNAc alignment may still be capable of homo-site binding, albeit at the cost of strain. However, an increased cross-site binding probability for large distances provides compensation, which might explain why the dissociation constants increase only moderately upon increase of the LacNAc–LacNAc spacing.
Molecular dynamics (MD) simulations were carried out in order to investigate the influence of nick sites and unpaired regions on the flexibility of the nucleic acid structure in more detail. For this purpose, we modeled complexes 18, 24 and 28, as they possess an equal number of bases between the LacNAc ligands. Complex 28 shares the same sequence as complex 18 but has a continuous PNA backbone without nick sites. The MD simulation of the complexes in water was started from the linear conformation. After an initial 10 ns equilibration time the simulation was extended for 30 ns in total. Though the simulation time probably is too short to realistically sample the conformational space of such large complexes, we assumed that differences in flexibility would be revealed.
The most rigid complex (28) showed a mean LacNAc–LacNAc distance that hardly differed from the estimated, canonical distance of 104 Å (Table 2). For the flexible complexes 18 and 24 we saw a deviation of about 5 Å from the predicted value. Whereas the probability for a bent conformation, i.e. a shorter LacNAc–LacNAc distance, increases for both complexes, only complex 18 shows a smaller LacNAc–LacNAc distance. Due to the increased base to base distance and the decreased helix diameter, the single strand regions of complex 24 expand and thereby compensate the distance decrease caused by the increased bending probability.



The MD simulations suggested differences in the dispersion of the torsion angle between the LacNAc presentation sites. While for double stranded complexes 18 and 28 the angle is in the same range, we found a clearly different value for complex 24. This and the increased standard deviation of the torsional angle obtained for complex 24 shows that the single strand segment indeed confers a higher flexibility than double strands. The MD simulation also exposed the gain in torsional freedom observed when the three nick sites are introduced in the double strand (Table 2, compare 18 with 28).
In a second step, we evaluated to which extent nick sites and unpaired regions promote the formation of a bent conformation which is required for bivalent binding to ECL. As 30 ns of MD only led to moderate bending of the strands, we set up a simulation where a constant force acted on the strand so that the required LacNAc–LacNAc distance could be adjusted. After obtaining bent, distance-adjusted conformations for complexes 18, 24 and 28, we performed short MD simulations to measure the force necessary to remain in these potential binding (i.e. correct LacNAc–LacNAc distance) conformations (Fig. 4). These values give a measure of strand flexibility, as they can also be interpreted as the force that has to act on the strand to adopt the bent conformation. As expected, the highest force was determined for the duplex with continuous PNA backbone (complex 28). The introduction of nick sites (complex 18) or single strand regions (complex 24) increased torsional freedom and the ability to stretch as the force to keep the complexes in the bent conformation decreased (Fig. 5). This effect was slightly higher for single strand regions (complex 24). Hence, conformations suitable for bivalent binding are more likely to be observed for these complexes. In general, the observations from MD are in agreement with the results from the SPR experiments.
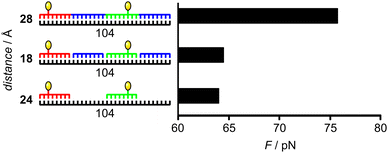 | ||
| Fig. 4 Mean force acting on the strand in the bent conformation with adjusted LacNAc–LacNAc distance, measured at the ligand attachment sites over 400 ps (see Table 1 for complex composition). | ||
 | ||
| Fig. 5 Conformations obtained by MD simulations in which a constant force acted on the strand to bend the complexes in a conformation suitable for bivalent binding to ECL. (a) Complex 18: bending occurs mainly at the nick sites while the duplex segments remain in a linear conformation. (b) Complex 24: the bent conformation is formed by a nick in the flexible unpaired region. (Images created with Amira;23 black = DNA strand, blue = PNA strands, green = linker). | ||
The purpose of this investigation was to explore whether programmed assembly of multipartite PNA–DNA complexes allows the interrogation of the distance between the binding sites of a lectin. In previous work, the recognition of DNA–galactose clusters by Ricinus communis agglutinin was studied. The valency of the glyco-assemblies that showed cooperative binding was not defined because the aggregates were prepared by polymerization through hybridization of half-sliding complementary oligonucleotides.7 We showed that the use of multiple, selectively addressable codon segments in DNA-templates (see ESI† for template sequences) enables a precise control of the number of presented glyco-ligands. Recent work has elegantly demonstrated that PNA–DNA hybridization can be used to dimerize multivalent high-mannose conjugates.8 The aim was to mimic the carbohydrate epitope of HIV. The high-mannose ligands were attached through oligoethyleneglycol linkers to the terminal ends of the PNA-oligomers. We preferred shorter linkers and the conjugation to internal positions to enable a better fine-tuning of ligand-ligand distances and to avoid blurring of distance constraints resulting from end-fraying. This, as well as the extension to multipartite PNA–DNA complexes and the consideration of partially unpaired template regions should facilitate the spatial screening of the distance between the carbohydrate binding sites of a lectin. Indeed, the presented work provides, to our best knowledge, the first example, in which self-organized supramolecular complexes have been used to unravel the arrangement of carbohydrate binding sites presented by a lectin.
Conclusions
In summary, we have prepared PNA-conjugates and demonstrated that hybridization with a DNA template allows the multivalent presentation of LacNAc ligands. A wide range of multivalent scaffolds was assembled by using only five different PNA oligomers and various DNA templates which contained selectively addressable codon sequences. We controlled both the number and the spatial arrangement of the LacNAc display through the combined use of multipartite PNA–DNA complexes and sequence internal glyco-conjugation, which would be difficult to achieve by using polymer-based scaffolds or nanoparticles. By introducing nick sites and unpaired regions into the PNA–DNA duplex the flexibility of the complexes could be altered, which is supported by MD simulations. The self-organized glyco-assemblies were used for the spatial screening of accessible carbohydrate binding sites in the Erythrina cristagalli lectin (ECL). This systematic investigation revealed a distance dependence which is in agreement with the crystal structure analysis. Though we report on the use of LacNAc as ligand, we believe that the permissive chemistry of PNA should allow the implementation of virtually any molecular structure. Current efforts are also directed to the multivalent display of peptide and steroid ligands.Acknowledgements
Support by the Deutsche Forschungsgemeinschaft (SFB 765) is most gratefully acknowledged. We thank Dr Sven Enders for help during the SPR experiments and Ole Schütt for help with the MD simulations.Notes and references
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 RSC.
- (a) G. B. Sigal, M. Mammen, G. Dahmann and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 3789–3800 CrossRef; (b) S.-K. Choi, M. Mammen and G. M. Whitesides, J. Am. Chem. Soc., 1997, 119, 4103–4111 CrossRef CAS; (c) D. D. Manning, X. Hu, P. Beck and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 3161–3162 CrossRef CAS.
- (a) K. Aoi, K. Itoh and M. Okada, Macromolecules, 1995, 28, 5391–5393 CrossRef CAS; (b) D. Zanini and R. Roy, J. Am. Chem. Soc., 1997, 119, 2088–2095 CrossRef CAS; (c) P. R. Ashton, E. F. Hounsell, N. Jayaraman, T. M. Nilsen, N. Spencer, J. F. Stoddart and M. Young, J. Org. Chem., 1998, 63, 3429–3437 CrossRef CAS.
- (a) J. M. de la Fuente, A. G. Barrientos, T. C. Rojas, J. Rojo, J. Cañada, A. Fernández and S. Penadés, Angew. Chem., Int. Ed., 2001, 40, 2257–2261 CrossRef; (b) Y. Chen, T. Ji and Z. Rosenzweig, Nano Lett., 2003, 3, 581–584 CrossRef CAS; (c) J. Zhang, C. D. Geddes and J. R. Lakowicz, Anal. Biochem., 2004, 332, 253–260 CrossRef CAS.
- (a) K. Matsuura, M. Hibino, Y. Yamada and K. Kobayashi, J. Am. Chem. Soc., 2001, 123, 357–358 CrossRef CAS; (b) K. Matsuura, M. Hibino, T. Ikeda, Y. Yamada and K. Kobayashi, Chem.–Eur. J., 2004, 10, 352–359 CrossRef CAS.
- K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS.
- E. Fan, Z. Zhang, W. E. Minke, Z. Hou, C. L. M. J. Verlinde and W. G. J. Hol, J. Am. Chem. Soc., 2000, 122, 2663–2664 CrossRef CAS.
- (a) P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS; (b) P. I. Kitov, H. Shimizu, S. W. Homans and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 3284–3294 CrossRef CAS.
- A. M. Riley, A. J. Laude, C. W. Taylor and B. V. L. Potter, Bioconjugate Chem., 2004, 15, 278–289 CrossRef CAS.
- N. Schaschke, G. Matschiner, F. Zettl, U. Marquardt, A. Bergner, W. Bode, C. P. Sommerhoff and L. Moroder, Chem. Biol., 2001, 8, 313–327 CrossRef CAS.
- (a) V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2000, 39, 4348–4352 CrossRef CAS; (b) V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 900–903 CrossRef CAS; (c) D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704–8719 CrossRef CAS.
- Z. Zhang, J. Liu, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Org. Chem., 2004, 69, 7737–7740 CrossRef CAS.
- K. Turton, R. Natesh, N. Thiyagarajan, J. A. Chaddock and K. R. Acharya, Glycobiology, 2004, 14, 923–929 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- E. Uhlmann, A. Peyman, G. Breipohl and D. W. Will, Angew. Chem., Int. Ed., 1998, 37, 2796–2823 CrossRef CAS.
- An adenine analogue has been used for native chemical ligation and conjugation: C. Dose and O. Seitz, Org. Lett., 2005, 7, 4365–4368 Search PubMed ; an Fmoc/StBu-protected thymine building block has been described: M. C. de Koning, L. Petersen, J. J. Weterings, M. Overhand, G. A. van der Marel and D. V. Filippov, Tetrahedron, 2006, 62, 3248–3258 CrossRef CAS.
- M. Eriksson and P. E. Nielsen, Nat. Struct. Biol., 1996, 3, 410–413 CrossRef CAS.
- E. Stenberg, B. Persson, H. Roos and C. Urbaniczky, J. Colloid Interface Sci., 1991, 143, 513–526 CrossRef CAS.
- X. Zeng, T. Murata, H. Kawagishi, T. Usui and K. Kobayashi, Carbohydr. Res., 1998, 312, 209–217 CrossRef CAS.
- J. C. Sacchettini, L. G. Baum and C. F. Brewer, Biochemistry, 2001, 40, 3009–315 CrossRef CAS.
- D. Stalling, M. Westerhoff and H.-C. Hege, in The Visualization Handbook, ed. C. D. Hansen and C. R. Johnson, Elsevier, 2005, ch. 38, pp. 749–767 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterizations, kinetic parameters of SPR experiments, and MD simulations. See DOI: 10.1039/c0sc00565g |
| This journal is © The Royal Society of Chemistry 2011 |