Switchable solvents
Pamela Polletac,
Charles A. Eckertabc and
Charles L. Liotta*abc
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: charles.liotta@chemistry.gatech.edu; Fax: +1 4048949085; Tel: +1 4048947070
bSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cSpecialty Separations Center, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 15th February 2011
Abstract
Switchable solvents are a unique class of solvents that were developed to facilitate both reaction and subsequent product separation. Their “built-in” separation ability for facile product recovery is paramount to achieving chemical processes that are both economically competitive and environmentally conscious. Two classes of switchable solvents are discussed: 1) piperylene sulfone—a volatile and recycle DMSO substitute and 2) one and two-component reversible ionic liquids—solvent systems that can be switched back and forth between molecular liquids and ionic liquids.
Introduction
The development of recyclable solvent systems that facilitate reaction and subsequent product separation is paramount to achieving chemical processes that are both economically competitive and environmentally conscious. The choice of the “right” solvent is critical not only for a successful chemical transformation but also for the subsequent separation and purification processes.1 For example, it is often desirable to react an ionic or polar reactant with a relatively non-polar reactant. Since the two reaction partners are not usually mutually soluble it is necessary to find a solvent medium that will partially or completely dissolve the two reactants in order to facilitate reaction to form the desired product. Under these circumstances dipolar, aprotic solvents such as dimethyl sulfoxide and dimethylformamide are often employed. Unfortunately, while the reaction may readily take place, the isolation of the product is often difficult because these solvents are high boiling and thus difficult to remove and cleanly separate from the product. In addition, the common procedures for isolating products from these expensive reaction solvents prohibit facile recycling of the solvent. In general, therefore, it is rarely practical to recycle and reuse such solvents. As a consequence, literature reports of the development of novel solvents are continually increasing. These solvent systems include ionic liquids,2 supercritical fluids,3 nearcritical liquids,3h,4 gas-expanded liquids,3h,5 organic-aqueous tuneable solvents (OATS)6 and perfluorinated solvents.7 This paper provides insights into potential chemical processes available for switchable solvents and the underlying physical properties that would make these processes possible. Its purpose is to provide the reader with an overview of the exciting opportunities provided by these novel reaction media and to show how these opportunities can be exploited for chemical processing, including both reactions and separations. Reversible ionic liquid systems have been reported in the literature which may not be describe herein,8 as this review is not intended to be comprehensive but rather presents illustrative examples.Switchable solvents are defined as solvents that reversibly change physical properties abruptly. This unique property is a consequence of a reversible reaction in response to an external stimulus such as a temperature change and/or the addition or removal of a gas. Because of the reversibility of the reaction, the changed solvent can easily be brought back to its original state. The possibility of reversibly “switching” the physical characteristics and/or the polarity of a solvent (ionic vs. covalent/liquid vs. gaseous) by controlled manipulation on a molecular level opens major paths for further basic research and future chemical process development.
The scope of this review is to highlight the potential of the emerging field of switchable solvents for a few representative applications. The focus will be placed on two classes of switchable solvents: 1) piperylene sulfone—a volatile and recyclable DMSO substitute and 2) one and two-component reversible solvent systems that can be switched back and forth between molecular liquids and ionic liquids.
Piperylene sulfone: “volatile” dipolar, aprotic solvent
Synthesis and properties of piperylene sulfone
Piperylene sulfone is synthesized from trans-1,3-pentadiene (trans-piperylene) and sulfur dioxide in the presence of a radical inhibitor (such as N-phenyl-2-naphtylamine).9 The synthesis can also be carried out using the commercially available and inexpensive 1,3-pentadiene, which is a mixture of trans- and cis- isomers. It should be noted that the cis-isomer reacts very slowly, if at all, with SO2 to form the cyclic sulfone. Vinci et al. measured and reported the solvatochromic properties of piperylene sulfone, showing that piperylene sulfone has comparable solvent properties to dimethyl sulfoxide (DMSO).10 The important difference between piperylene sulfone and DMSO is that the former is switchable and the latter is not. Indeed, at temperatures greater than 100 °C piperylene sulfonecleanly and efficiently decomposes to gaseous trans-1,3-pentadiene (b.p. 42 °C) and sulfur dioxide (b.p. −10 °C) via a retro-cheletropic process (Fig. 1). The products can be collected and allowed to react to reform the piperylene sulfone solvent.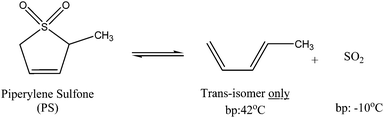 | ||
| Fig. 1 Reversible decomposition of piperylene sulfone into trans-piperylene and sulfur dioxide. | ||
Critical to employing piperylene sulfone as a solvent for coupling reactions and separations is the requirement that decomposition of the solvent occurs with 100% efficiency. Differential scanning calorimetry coupled with thermogravimetric analysis was performed on piperylene sulfone. Heating at 5 °C min−1 for 20 min and then holding at 120 °C for 30 min triggered the decomposition, leaving no residual mass. Thus, in principle, facile product recovery and solvent recycle can be realized. Piperylene sulfone is one of a number of possible β,γ-unsaturated cyclic sulfones (also called sulfolenes) that can be employed as solvents. Other sulfolenes such as butadiene sulfone and isoprene sulfone are also of interest for switchable solvents applications.11
Chemical processes in piperylene sulfone: Substitution reactions
Since piperylene sulfone is proposed as a substitute for dimethylsulfoxide, it was necessary to compare the two solvents under similar conditions in order to assess the advantages and disadvantages of piperylene sulfone. Vinci et al. reported the rates of reaction of benzyl chloride with a variety of anionic nucleophiles in both piperylene sulfone and DMSO at 40 °C.10 The results are summarized in Table 1. The authors discovered that surprisingly traces of water added to the piperylene sulfone affect the rate of reaction of several anionic nucleophiles. For instance, no reaction was observed with potassium cyanide in anhydrous piperylene sulfone. Only in the presence of traces of water (0.1 wt%) did the reaction take place. However it was found that the addition of more water (up to 10%) did not affect significantly the reaction rate further. In most cases the rates of reaction were only slightly greater in the presence of small quantities of water. For example, the nucleophiles potassium thiocyanate and potassium acetate showed a rate enhancement in the presence of small quantities of water in both DMSO and piperylene sulfone. As a consequence, studies were conducted in dry DMSO, DMSO containing 3 wt% water (commercial reagent grade), dry piperylene sulfone, and piperylene sulfone containing 1 wt% water (Table 1).† In general, the reactions in both solvents are quantitative and the rates are slower in piperylene sulfone compared with DMSO.
|
||||
|---|---|---|---|---|
| 2nd Order Rate Constant k × 101 (mL mol−1s−1) | ||||
| Nucleophile | DMSO | DMSO (3% H2O) | Piperylene Sulfone | Piperylene sulfone (1% H2O) |
| a KTA: potassium thioacetate, NaPDTC: Sodium pyrrolidinedithiocarbamate.10 | ||||
| KTA a | >1800 | >1800 | >1800 | >1800 |
| NaPDTC a | >1800 | >1800 | >1800 | >1800 |
| KSCN | 1.4 (±0.1) | 1.7 (±0.1) | 2.1 (±0.1) | 2.3 (±0.2) |
| KOAc | 3.4 (±0.1) | 11.0 (±0.1) | 0.013 (±0.004) | 0.19 (±0.01) |
| KCN | 5.8 (±0.8) | 17 (±5) | No rxn | 0.15(±0.01) |
| CsN3 | 69 (±1) | 16.7 (±0.9) | 2.4 (±0.9) | 5.8 (±0.9) |
| CsOAc | 22.7 (±0.6) | 16.4 (±0.2) | 0.35 (±0.04) | 0.35 (±0.06) |

With the fundamental reaction parameters in hand, the actual process, which includes reaction, solvent removal, product isolation, and solvent regeneration and recycle, can be outlined as seen in Fig. 2. After the reaction of benzyl chloride with potassium thiocyanate in piperylene sulfone at 40 °C, the product reaction mixture was heated to 110 °C. This initiated the decomposition of the solvent to gaseous trans-piperylene and sulfur dioxide, leaving behind the solvent-free product residue. The pure benzyl thiocyanate was isolated from the residue in 96% yield. The gases from the solvent decomposition were collected in a second vessel containing liquid sulfur dioxide at −30 °C. After warming up to room temperature, sulfur dioxide and trans-piperylene were left to react. Piperylene sulfone was quantitatively reformed and isolated. Piperylene sulfone was isolated in 87% yield. Although the recovery is relatively good, it is lower than one would want for a process. This low recovery is due to minor losses because of the small scale of the laboratory procedure – simple surface adhesion or adsorption accounts for most of these losses. Would the process be scaled up, the percent recovery would undoubtedly be far greater.
 | ||
| Fig. 2 Schematic illustrations of the four stage process: 1) Reaction, 2) solvent decomposition/product isolation, 3) solvent reformation and 4) solvent recycling.10 | ||
Reversible ionic liquids
A reversible ionic liquid is a solvent system which can switch back and forth between a medium which is ionic and a medium which is non-ionic; the latter is referred to as a molecular medium. There are two major classes of reversible ionic liquids (RevILs): two-component and one-component.(1) Two-component RevILs are based on an equimolar mixture of a neutral alcohol and a neutral molecule containing at least one basic nitrogen functionality. Examples of the latter are 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 2-butyl-1,1,3,3-tetramethylguanidine (TMBG). Upon exposure to carbon dioxide the alcohol forms an alkyl carbonic acid which subsequently transfers a proton to the basic nitrogen to form an ionic medium. The process is easily reversed to reform the alcohol and the basic nitrogen compound by simply warming the system or sparging with an inert gas. The overall reversible process is shown in Fig. 3. It should be emphasized that maintaining an equimolar mixture of the guanidine or amidine and the alcohol throughout a chemical process could be difficult—especially when dealing with low boiling point alcohols such as methanol.
![Reversible switch from a molecular liquid mixture of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or N,N,N′,N′-tetramethyl-N′′-butylguanidine (TMBG) and alcohol (ROH) to the ionic liquid [DBUH]+ [RCO3]− and [TMBGH]+[RCO3]− upon addition of CO2, respectively. R = C1 to C12.](/image/article/2011/SC/c0sc00568a/c0sc00568a-f3.gif) | ||
| Fig. 3 Reversible switch from a molecular liquid mixture of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or N,N,N′,N′-tetramethyl-N′′-butylguanidine (TMBG) and alcohol (ROH) to the ionic liquid [DBUH]+ [RCO3]− and [TMBGH]+[RCO3]− upon addition of CO2, respectively. R = C1 to C12. | ||
Conventional ionic liquids (ILs) have been used as solvent vehicles for a wide variety of chemical reactions. In contrast to RevIL's conventional ionic liquids remain ionic throughout the course of reaction and separation. As reactions occur readily, and many times in high yields, the isolation of the product is often difficult. Because of the switchable property of RevIL's many of the product isolation problems encountered by conventional IL's can be circumvented.11 Like conventional ionic liquids (ILs), reversible ionic liquids (RevILs) can be designed for a specific application, where the molecular architecture of the ionic solvent system can be adjusted to achieve the desired solvent properties.
(2) One-component systems eliminate the need for an alcohol as seen in the two-component RevILs; these systems employ solely a neutral molecule containing at least one basic nitrogen functionality.12 An example of a one-component system are where the silylated amines such as trialkoxy- or trialkyl-silylpropylamines are used as the molecular liquid. Upon reaction with CO2, these amine precursors form ionic liquids composed of the corresponding carbamate anion and ammonium cation pairs (Fig. 4). The Lewis acid character of the silicon substituent is believed to play a role in controlling the temperatures at which carbon dioxide is captured and released.
 | ||
| Fig. 4 One-component system: reversible switch from a molecular liquid trialkoxy- and trialkyl-silylpropylamine to its corresponding ionic liquid upon addition of CO2. | ||
The synthesis and characterization of both classes of reversible ionic liquids (two-component and one-component) is briefly reviewed. This will be followed by applications of RevILs coupling reactions and separations. Examples presented here are (1) the Heck reaction in which the reaction is conducted in the ionic liquid and (2) the Claisen-Schmidt condensation in which the reaction is conducted in the molecular liquid.
Physical properties and synthesis of two and one component ionic liquids
Two and one component RevILs are generated by bubbling CO2 through equimolar solutions of basic nitrogen functional molecules and various primary alcohols (from methanol to dodecanol) or through neat basic nitrogen functional molecules, respectively. Reversal of the resulting ionic liquid back to the molecular liquid is achieved by bubbling N2 (or other inert gases) through the ionic liquid and/or mild heating (60 °C). The reversible formation of the ionic products can be characterized by 1H and 13C NMR, elemental analysis, FT-IR, melting points, differential scanning calorimetry (DSC), and conductivity measurement.The relative polarities of the molecular liquid and the corresponding ionic liquids can be determined viaUV-Vis absorption measurement of the Nile Red dye (a common solvatochromic probe), see Table 2.12
| Solvent | lmax (nm), Nile Red |
|---|---|
| Ether | 504 |
| CH2Cl2 | 535 |
| CHCl3 | 538 |
| TMBG + MeOH | 538 |
| DBU + MeOH | 538 |
| DMF | 541 |
| Propanoic acid | 542 |
| [bmim]+[PF6]− | 548 |
| [DBUH]+[MeOCO2]− | 548 |
| DMSO | 549 |
| [bmim]+[PF6]− | 551 |
| [TMBGH]+[MeOCO2]− | 554 |
| Acetic acid | 557 |
Equimolar mixtures of DBU/alcohol or TMBG/alcohol become significantly more polar when exposed to CO2, as shown by the shift of the λmax to longer wavelengths. For example, the TMBG/methanol mixture exhibits a λmax of 538.0 nm while the λmax of the corresponding ionic liquid (N,N,N′,N′-tetramethyl-N′′-butylguanidinium ethylcarbonate) is 554.0 nm, corresponding to a shift of 16.0 nm. Such a shift in λmax represents a polarity switch akin to going from chloroform to acetic acid. The Nile Red experiments suggest that the polarity of both molecular and ionic forms depend on the length of the alkyl chain on the alcohol. The many possible combinations of base and alcohol give a wide selection of solvent switches. The magnitude in change of polarity for one-component trialkyl and trialkoxysilylamine-based RevILs is about 10 nm, which is less than for some of the two-component solvents.
It should be noted that upon reversal to the molecular liquid, the two-component systems which utilize low molecular weight alcohols, such as methanol, show the concurrent loss of the alcohol and CO2.13 In contrast, the loss of CO2 for the one-component system occurs at a temperature substantially different (<100 °C) from the evaporation of the molecular liquid—an advantage in term of ease of processing and ionic liquid preparation. Because of the Si–O bond, the trialkoxy-silylpropylamine system has limited stability to water (cleavage of the Si–O bond occurs readily). This limitation does not extend to trialkyl-silylpropylamines (the Si–C bond cannot be hydrolyzed). This is an important consideration for the application of the one-component RevIL solvents to scenarios where water is present, required or produced.
The Heck reaction
Heck reactions of bromobenzene with styrene in the presence of Pd-catalysts were investigated in the two-component reversible ionic liquid mixture of DBU/hexanol. The overall process was designed to couple the reaction, which was carried out under ionic conditions, and a two-stage separation: first isolating the nonpolar product from the ionic liquid, and second by precipitating the salt by-product from the nonpolar solvent mixture (upon “switching” the ionic liquid). In principle, the reversible “switch” from ionic to molecular solvent should enable the separation of product and by-product sequentially, leading to a recycling of the solvent system and the catalyst. As a benchmark reaction, the palladium catalyzed Heck reaction of bromobenzene and styrene was investigated (Fig. 5). E-stilbene was produced as the major product. Other isomers (1,1-diphenylethylene and Z-stilbene) were obtained in yields of less than 5%. Hart et al. found that the complex PdCl2(PPh3)2 prepared ex-situ gave the best performance, with up to a 97% yield of E-stilbene using a catalyst loading of 2 mol%.14 No additional base was required in these experiments since free DBU, in equilibrium with the ionic species, was found to act as a scavenger for HBr.![Heck reaction of bromobenzene and styrene in the reversible ionic liquid [DBUH]+[HexOCO2]−.14](/image/article/2011/SC/c0sc00568a/c0sc00568a-f5.gif) | ||
| Fig. 5 Heck reaction of bromobenzene and styrene in the reversible ionic liquid [DBUH]+[HexOCO2]−.14 | ||
Extraction of the products from the reaction mixture was carried out under a CO2 atmosphere to ensure that [DBUH]+[HexOCO2]− remained in the ionic form. However, GC analysis indicated that some free DBU and hexanol were also extracted into the heptane phase. After phase separation, the ionic phase was reverted back to the DBU/hexanol molecular liquid. As a consequence, the colorless HBr salt of DBU separated from the solution and was easily isolated by filtration. The supernatant solution remained yellow, suggesting the presence of dissolved palladium catalyst.
The application of RevILs to the Heck reaction offers an effective solution for the problem of salt accumulation. The reaction was carried out in the ionic liquid form of the solvent, from which the nonpolar product could be isolated by extraction. Reversing the solvent to its molecular liquid state caused the salt by-product to precipitate. After separation of the salt and addition of fresh DBU to compensate for losses, the solvent (containing the catalyst) was switched back to its ionic form and was used for another Heck reaction showing significant activity.
The Claisen-Schmidt condensation
The Claisen-Schmidt condensation of butanone and benzaldehyde in RevIL system (TMBG/MeOH) was reported by Hart et al. (Fig. 6). Three products are formed upon reaction: the internal enone (3-methyl-4-phenyl-but-3-en-2-one), the terminal enone (1-phenyl-pent-1-en-3-one) and water.15 Under basic conditions, the terminal enone product is the predominant product formed.16 It should noted that TMBG plays the dual role of base catalyst and solvent. | ||
| Fig. 6 Claisen-Schmidt condensation of 2-butanone and benzaldehyde. | ||
After 24 h at room temperature or 3 h at 80 °C, yields of 48% and 44% in enone products were obtained, respectively. The formation of the terminal and internal enone-products was also studied as a function of time at 80 °C. As the reaction time increased, the yield of enone products first increased from 13% at 1 h to 44% at 3 h and then began to decrease after 4 h. The decrease was attributed to competing condensation processes between the enone products and the benzaldehyde, resulting in lower overall yields of the desired products. As a consequence, shorter reaction times to partial conversions were necessary in order to develop a process in which isolated yields were maximized and solvent recycle was possible (Fig. 7).
 | ||
| Fig. 7 Process that couples reaction and separation for the Claisen-Schmidt condensation of butanone and benzaldehyde. | ||
The isolation of the enone products was performed by adding n-octane and methanol to the reaction mixture, followed by the addition of CO2, which triggered the formation of the ionic liquid. Under these conditions an octane phase separated from the newly developed ionic liquid phase. The enone products were predominantly soluble in the octane phase and were easily separated by decantation.
It should be pointed out that water, a product from the condensation reaction, can react with TMBG and CO2 to form the N,N,N′,N′-tetramethyl-N′′-butylguanidium carbonate, preventing the ionic liquid reversal and recycling. The successful reversal of the ionic liquid and recycle of TMBG was however demonstrated when the reaction mixture was dried with magnesium sulfate (and filtered) prior to reaction with CO2 (Fig. 7). By introducing the drying step into the process, the authors showed that TMBG could be recycled three times. The isolated yields of enone products were 34%, 32% and 34% for each cycle with a consistent product distribution of 95% terminal enone product. Again, it should be emphasized that partial conversion was necessary in order to avoid the higher condensation products, thus providing recycling for the Claisen-Schmidt condensation in RevILs.
Conclusions
Solvents are necessary for most chemical processes and lead to a large percentage of the process cost and to the size of the waste-stream. In order to achieve chemical processes that are both competitive and environmentally conscious, solvents must address simultaneously reaction efficiency, product separation, and recyclability. Switchable solvents were developed to meet these criteria. Piperylene sulfone has been demonstrated to be a good medium for reactions of anionic nucleophiles with organic electrophiles, oxidations of alcohols to aldehydes17 and in situ reversible acid-catalyzed hydrolysis.18 It is a dipolar, aprotic solvent which mimics the properties of DMSO. However, unlike DMSO, because of a facile and reversible reaction between trans-piperylene and sulfur dioxide, product isolation and solvent recycle are easily accomplished. In addition, Heck coupling and Claisen-Schmidt condensation have been successfully carried out in RevILs with the added capabilities of improved separation of the product and recycling of the solvent system. Reversible ionic liquids facilitate catalyst and product recovery, yielding efficient separations of catalysts for reuse. Switchable solvents are one class of innovative solvent systems that clearly demonstrate that solvents can accomplish more than heat and mass transfer; they can actively contribute to facilitate reaction and product separation while minimizing waste generation.Notes and references
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry. 3rd ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, 2003p653 Search PubMed.
- (a) R. D. Rogers; K. R. Seddon, Ionic Liquids as Green Solvents: Progress and Prospects. [In: ACS Symp. Ser., 2003; 856]. American Chemical Society: Washington, D.C., 2003; p 599 Search PubMed; (b) P. Wasserscheid; T. Welton, Ionic Liquids in Synthesis. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim: 2003 Search PubMed.
- (a) R. L. Thompson, R. Glaser, D. Bush, C. L. Liotta and C. A. Eckert, Rate variations of a hetero-Diels–Alder reaction in supercritical fluid CO2, Ind. Eng. Chem. Res., 1999, 38(11), 4220–4225 CrossRef CAS; (b) J. S. Brown, H. P. Lesutis, D. R. Lamb, D. Bush, K. Chandler, B. L. West, C. L. Liotta, C. A. Eckert, D. Schiraldi and J. S. Hurley, Supercritical fluid separation for selective quaternary ammonium salt promoted esterification of terephthalic acid, Ind. Eng. Chem. Res., 1999, 38(10), 3622–3627 CrossRef CAS; (c) A. K. Dillow, J. S. Brown, C. L. Liotta and C. A. Eckert, Supercritical fluid tuning of reactions rates: The cis-trans isomerization of 4-4′-disubstituted azobenzenes, J. Phys. Chem. A, 1998, 102(39), 7609–7617 CrossRef CAS; (d) G. Brunner, Supercritical Fluids as Solvents and Reaction Media. Elsevier B.V.: Amsterdam, 2004; p 641 Search PubMed; (e) P. G. Jessop; W. Leitner, Chemical Synthesis Using Supercritical Fluids. WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, 1999; p 480 Search PubMed; (f) R. A. Brown, P. Pollet, E. McKoon, C. A. Eckert, C. L. Liotta and P. G. Jessop, Asymmetric hydrogenation and catalyst recycling using ionic liquid and supercritical carbon dioxide, J. Am. Chem. Soc., 2001, 123(6), 1254–1255 CrossRef CAS; (g) S. A. Nolen, J. Lu, J. S. Brown, P. Pollet, B. C. Eason, K. N. Griffith, R. Glaser, D. Bush, D. R. Lamb, C. L. Liotta, C. A. Eckert, G. F. Thiele and K. A. Bartels, Olefin epoxidations using supercritical carbon dioxide and hydrogen peroxide without added metallic catalysts or peroxy acids, Ind. Eng. Chem. Res., 2002, 41(3), 316–323 CrossRef CAS; (h) C. A. Eckert, C. L. Liotta, D. Bush, J. S. Brown and J. P. Hallett, Sustainable reactions in tunable solvents, J. Phys. Chem. B, 2004, 108(47), 18108–18118 CrossRef CAS; (i) D. Koch and W. Leitner, Rhodium-catalyzed hydroformylation in supercritical carbon dioxide, J. Am. Chem. Soc., 1998, 120(51), 13398–13404 CrossRef CAS; (j) A. Furstner, L. Ackermann, K. Beck, H. Hori, D. Koch, K. Langemann, M. Liebl, C. Six and W. Leitner, Olefin metathesis in supercritical carbon dioxide, J. Am. Chem. Soc., 2001, 123(37), 9000–9006 CrossRef CAS; (k) M. Solinas, J. Y. Jiang, O. Stelzer and W. Leitner, A cartridge system for organometallic catalysis: Sequential catalysis and separation using supercritical carbon dioxide to switch phases, Angew. Chem., Int. Ed., 2005, 44(15), 2291–2295 CrossRef CAS; (l) G. Maayan, B. Ganchegui, W. Leitner and R. Neumann, Selective aerobic oxidation in supercritical carbon dioxide catalyzed by the H5PV2Mo10O40 polyoxometalate, Chem. Commun., 2006,(21), 2230–2232 RSC.
- (a) K. Chandler, F. H. Deng, A. K. Dillow, C. L. Liotta and C. A. Eckert, Alkylation reactions in near-critical water in the absence of acid catalysts, Ind. Eng. Chem. Res., 1997, 36(12), 5175–5179 CrossRef CAS; (b) K. Chandler, C. L. Liotta, C. A. Eckert and D. Schiraldi, Tuning alkylation reactions with temperature in near-critical water, AIChE J., 1998, 44(9), 2080–2087 CAS; (c) H. P. Lesutis, R. Glaser, C. L. Liotta and C. A. Eckert, Acid/base-catalyzed ester hydrolysis in near-critical water, Chem. Commun., 1999,(20), 2063–2064 RSC; (d) H. R. Patrick, K. Griffith, C. L. Liotta, C. A. Eckert and R. Glaser, Near-critical water: A benign medium for catalytic reactions, Ind. Eng. Chem. Res., 2001, 40(26), 6063–6067 CrossRef CAS; (e) S. A. Nolen, C. L. Liotta, C. A. Eckert and R. Glaser, The catalytic opportunities of near-critical water: a benign medium for conventionally acid and base catalyzed condensations for organic synthesis, Green Chem., 2003, 5(5), 663–669 RSC.
- (a) J. P. Hallett, C. L. Kitchens, R. Hernandez, C. L. Liotta and C. A. Eckert, Probing the Cybotactic Region in Gas-Expanded Liquids (GXLs), Acc. Chem. Res., 2006 Search PubMed in press ; (b) A. M. Scurto, E. Newton, R. R. Weikel, L. Draucker, J. Hallett, C. L. Liotta, W. Leitner and C. A. Eckert, Melting point depression of ionic liquids with CO2: Phase equilibria, Ind. Eng. Chem. Res., 2008, 47(3), 493–501 CrossRef CAS; (c) P. G. Jessop, R. R. Stanley, R. A. Brown, C. A. Eckert, C. L. Liotta, T. T. Ngo and P. Pollet, Neoteric solvents for asymmetric hydrogenation: supercritical fluids, ionic liquids, and expanded ionic liquids, Green Chem., 2003, 5(2), 123–128 RSC; (d) T. S. Chamblee, R. R. Weikel, S. A. Nolen, C. L. Liotta and C. A. Eckert, Reversible in situ acid formation for beta-pinene hydrolysis using CO2 expanded liquid and hot water, Green Chem., 2004, 6(8), 382–386 RSC; (e) X. F. Xie, C. L. Liotta and C. A. Eckert, CO2-catalyzed acetal formation in CO2-expanded methanol and ethylene glycol, Ind. Eng. Chem. Res., 2004, 43(11), 2605–2609 CrossRef CAS; (f) R. R. Weikel, J. P. Hallett, C. L. Liotta and C. A. Eckert, Self-neutralizing in situ acid catalysis for single-pot synthesis of iodobenzene and methyl yellow in CO2-Expanded methanol, Ind. Eng. Chem. Res., 2007, 46(16), 5252–5257 CrossRef CAS; (g) J. P. Hallett, J. W. Ford, R. S. Jones, P. Pollet, C. A. Thomas, C. L. Liotta and C. A. Eckert, Hydroformylation catalyst recycle with gas-expanded liquids, Ind. Eng. Chem. Res., 2008, 47(8), 2585–2589 CrossRef CAS; (h) P. G. Jessop and B. Subramaniam, Gas-expanded liquids, Chem. Rev., 2007, 107(6), 2666–2694 CrossRef CAS.
- Pamela Pollet, R. J.H., Charles A. Eckert and Charles. L. Liotta, Organic Aqueous Tunable Solvents (OATS): A Vehicle for Coupling Reactions and Separations, Accounts Chem Res, 2010 Search PubMed ASAP .
- (a) C. D. Ablan, J. P. Hallett, K. N. West, R. S. Jones, C. A. Eckert, C. L. Liotta and P. G. Jessop, Chem. Commun., 2003, 2972 RSC; (b) Y. W. Kho, D. C. Conrad, R. A. Shick and B. L. Knutson, Ind. Eng. Chem. Res., 2003, 42, 6511 CrossRef CAS; (c) M. S. Yu, D. P. Curran and T. Nagashima, Org. Lett., 2005, 7, 3677 CrossRef CAS.
- (a) L. Phan, J. R. Andreatta, L. K. Horvey, C. F. Edie, A. L. Luco, A. Mirchandani, D. J. Darensbourg and P. G. Jessop, Switchable-polarity solvents prepared with a single liquid component, J. Org. Chem., 2008, 73(1), 127–132 CrossRef CAS; (b) T. Yu, T. Yamada, G. C. Gaviola and R. G. Weiss, Carbon Dioxide and Molecular Nitrogen as Switches between Ionic and Uncharged Room-Temperature Liquids Comprised of Amidines and Chiral Amino Alcohols, Chem. Mater., 2008, 20(16), 5337–5344 CrossRef CAS; (c) T. Yamada, P. J. Lukac, M. George and R. G. Weiss, Reversible, room-temperature ionic liquids. Amidinium carbamates derived from amidines and aliphatic primary amines with carbon dioxide, Chem. Mater., 2007, 19(5), 967–969 CrossRef CAS; (d) T. Yamada, P. J. Lukac, T. Yu and R. G. Weiss, Reversible, room-temperature, chiral ionic liquids. amidinium carbamates derived from amidines and amino-acid esters with carbon dioxide, Chem. Mater., 2007, 19(19), 4761–4768 CrossRef CAS; (e) J. C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A. C. Gaumont, Ionic Liquids: New Targets and Media for alpha-Amino Acid and Peptide Chemistry, Chem. Rev., 2008, 108(12), 5035–5060 CrossRef CAS.
- (a) O. Grummitt, A. E. Ardis and J. Fick, Thermal Dissociation of Methyldihydrothiophene-1-Dioxides, J. Am. Chem. Soc., 1950, 72(11), 5167–5170 CrossRef CAS; (b) G. A. Marus, E. Vyhmeister, P. Pollet, M. E. Donaldson, V. Llopis-Mestre, S. Faltermeier, R. Roesel, L. Gelbaum, C. L. Liotta and C. E. Eckert, The sustainable and Scalable Synthesis of Piperylene Sulfone: A “Volatile” and Recyclable DMSO Substitute, Ind Eng Chem Res, 2010 Search PubMed In Press.
- D. Vinci, M. Donaldson, J. P. Hallett, E. A. John, P. Pollet, C. A. Thomas, J. D. Grilly, P. G. Jessop, C. L. Liotta and C. A. Eckert, Piperylene sulfone: a labile and recyclable DMSO substitute, Chem. Commun., 2007,(14), 1427–1429 RSC.
- L. R. Drake, S. C. Stowe and A. M. Partansky, Kinetics of the Diene Sulfur Dioxide Reaction, J. Am. Chem. Soc., 1946, 68(12), 2521–2524 CrossRef CAS.
- L. Phan, D. Chiu, D. J. Heldebrant, H. Huttenhower, E. John, X. W. Li, P. Pollet, R. Y. Wang, C. A. Eckert, C. L. Liotta and P. G. Jessop, Switchable solvents consisting of amidine/alcohol or guanidine/alcohol mixtures, Ind. Eng. Chem. Res., 2008, 47(3), 539–545 CrossRef CAS.
- V. Blasucci, R. Hart, V. L. Mestre, D. J. Hahne, M. Burlager, H. Huttenhower, B. J. R. Thio, P. Pollet, C. L. Liotta and C. A. Eckert, Single component, reversible ionic liquids for energy applications, Fuel, 2010, 89(6), 1315–1319 CrossRef CAS.
- R. Hart, P. Pollet, D. J. Hahne, E. John, V. Llopis-Mestre, V. Blasucci, H. Huttenhower, W. Leitner, C. A. Eckert and C. L. Liotta, Benign coupling of reactions and separations with reversible ionic liquids, Tetrahedron, 2010, 66(5), 1082–1090 CrossRef CAS.
- (a) S. Nolen, C. L.L., C. A. Eckert and R. Glaser, The catalytic opportunities of near-critical water: a benign medium for conventionally acid and base catalyzed condensations in organic synthesis, Green Chem., 2003, 5, 663–669 RSC; (b) F. A. C.a. R. J. Sunberg, Advanced Organic Chemistry, Part B: Reactions and Synthesis. 3rd ed ed.; Plenum Press: New York, NY, 1990 Search PubMed.
- M. Stiles, D. Wolf and G. V. Hudson, Catalyst Selectivity in the Reactions of Unsymmetrical Ketones - Reaction of Butanone with Benzaldehyde and Para-Nitrobenzaldehyde, J. Am. Chem. Soc., 1959, 81(3), 628–632 CrossRef CAS.
- N. Jiang, D. Vinci, C. L. Liotta, C. A. Eckert and A. J. Ragauskas, Piperylene sulfone: A recyclable dimethyl sulfoxide substitute for copper-catalyzed aerobic alcohol oxidation, Ind. Eng. Chem. Res., 2008, 47(3), 627–631 CrossRef CAS.
- M. E. Donaldson, V. L. Mestre, D. Vinci, C. L. Liotta and C. A. Eckert, Switchable Solvents for in situ Acid-Catalyzed Hydrolysis of beta-Pinene, Ind. Eng. Chem. Res., 2009, 48(5), 2542–2547 CrossRef CAS.
Footnote |
| † The quantities of water in each of the solvent systems were determined by standard Karl-Fischer titrations. |
| This journal is © The Royal Society of Chemistry 2011 |