Coordination chemistry of group 13 monohalides
Dragoslav
Vidovic
and
Simon
Aldridge
*
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: Simon.Aldridge@chem.ox.ac.uk; Fax: +44 (0)1865 272690; Tel: +44 (0)1865 285201
First published on 8th February 2011
Abstract
The Group 13 monohalides, EX, are valence isoelectronic with the key textbook diatomic molecules CO and N2; recent advances in synthetic chemistry leading to the isolation of transition metal complexes containing such fragments as ligands are appraised, together with studies of the electronic structure and reactivity of the coordinated molecules.
Introduction
Traditionally, the chemistry of the Group 13 elements has been dominated by compounds in the +3 oxidation state, with applications exploiting their inherent Lewis acidity, for example, in catalysis, sensing, and more recently in frustrated Lewis pairs.1,2 Research into systems featuring lower formal oxidation states has typically been dominated by borane clusters and - given the tenets of the ‘Inert Pair Effect’ - by the extended ionic solids of In(I) and Tl(I).1 More recently a wider range of subvalent systems have become available, reflecting not only the use of certain reagents (especially sub-valent indium derivatives) in organic synthesis,3 but also the development of ground-breaking synthetic approaches. For example, the work of Schnöckel has allowed access to metastable Al(I) and Ga(I) halides, by making use of the fact that the equilibrium defined by eqn (1) can be driven to the right on entropic grounds at higher temperatures (E = B–Tl).4–6| EX3(g) + 2E(s or 1) ⇄ 3EX(g) | (1) |
Subsequent trapping and derivatization has allowed access to a wide range of subvalent aluminium and gallium compounds, including remarkable nano-sized clusters, the formation of which is suggested by theory to mimic the aggregation of related phases of the elemental metals.5 Much chemistry in solution has exploited sterically bulky substituents (e.g.Cp*, terphenyl, amino, β-diketiminate, guanidinate groups) as a strategy for isolating discrete molecular systems.7 Moreover, the ability of E(I) systems bearing such substituents to act as ligands towards transition metal centres has been widely established, particularly for boron, aluminium and gallium.7 By contrast, the coordination chemistries of the simple Group 13 monohalides have remained, until recently, a largely uncharted area, despite the valence isoelectronic relationship between such species and the ubiquitous textbook diatomic molecules CO and N2. While the comparative electronic structures, and consequent properties as ligands, of BF, CO and N2, for example, have been thoroughly examined computationally,8 complementary experimental data have remained stubbornly absent. Such deficiencies naturally reflect the low steric loading and high polarity of the coordinated EX fragment (as predicted computationally). Recent synthetic advances aimed at circumventing these problems for EX and related ligand systems are detailed in this mini-review.
Boron monohalides
The ‘free’ molecules
Since the first reports of its properties appeared in 1935, the gaseous boron monofluoride molecule has been the subject of numerous spectroscopic and thermodynamic studies,9–23 employing, for example, high temperature in situ synthesis from calcium fluoride and elemental boron. Thus, an equilibrium bond distance (re) of 1.26267(1) Å has been determined from microwave spectroscopy,22 and values also determined for Do (757 ± 14 kJ mol−1),23ωe (1765 ± 20 cm−1),20 and first ionization potential, I1 (11.115 ± 0.004 eV from electronic absorption spectra).16 In a similar fashion, bond lengths for the heavier halide diatomics BX have also been determined [X = Cl: 1.715, Br: 1.888, I: 2.131 Å].1By contrast, the synthesis of BF on a preparative scale was not reported until 1967, relying on the seminal work of Peter Timms to develop a comproportionation based synthetic route from BF3 and elemental boron.24–26 This synthesis requires a specially built reactor in which gaseous BF3 is passed over elemental boron at reduced pressure (< 1 mmHg) and elevated temperature (ca. 2000 °C) to generate the product in ca. 85% yield.24 Moreover, it is apparent that, while boron monofluoride retains its diatomic nature in the vapour phase, condensation at −196 °C leads to the formation of a polymeric material. Such behaviour is not entirely unexpected, given the oligomeric structures known for related B(I) compounds such as (tBuB)4, (tmpB)4 and (XB)9 (X = Cl, Br).27 Subsequent warming leads to the formation of a number of B/F containing compounds (including B2F4, B3F5 and B8F12),24,28,29 the proportions of which reflect the amount of BF3 co-condensate. Thus B2F4 is formed by BF insertion into BF3, and B3F5 by subsequent insertion of BF into B2F4.24 Timms also reported the synthesis of metastable boron monochloride, which can be achieved by the controlled, high temperature, low pressure cracking of B2Cl4 (yielding BCl and BCl3) or by the reaction of BCl3 with elemental boron at 2000 °C.26,30
Matrix isolation provides a viable method for the trapping and interogation of the BX diatomics, although the synthetic route employed, utilizing the reaction between laser ablated boron atoms and the respective X2 molecules, is not selective; mixtures of BX3, BX2 and BX are generated for all four halogens. Infrared spectroscopy yields vibrational frequencies for the BX molecules isolated in solid argon of 1374 (11BF), 815/810 (11B35Cl/11B37Cl), 667/666 (11B79Br/11B81Br) and 564 cm−1 (11BI).31
Reactivity-wise, initial studies suggested that BF insertion, although facile for B–F bonds, is less effective with other E–F linkages; the analogous reactions with C2F4 and SiF4 resulted in the synthesis of only trace amounts of insertion products. In the presence of soft donors (e.g.CO, PF3, PCl3 PH3, AsH3 and SMe2) condensation of BF leads to the formation of compounds of the general formula (F2B)3B·L, with the carbonyl complex having been characterized crystallographically;32 related chemistry has also been reported for BCl.30 The reactions of BF with unsaturated hydrocarbons such as acetylene or propene lead to the formation of acyclic and cyclic products incorporating BF or BF2 moieties,25 while the analogous reactions with gaseous BCl lead to the exclusive formation of cyclic compounds. Related chemistry with photolytically generated Ph3SiB leads to the formation of a borirene heterocycle with acetylene (viacycloaddition), and to insertion products with alkanes and with THF.33
Coordination chemistry
The possibility of using boron monohalides as ligands in transition metal coordination complexes was investigated by quantum chemical methods in a series of papers published in 1998.8 In particular, the comparative bonding properties of the 10 valence electron diatomics N2, CO and BF were appraised. The relative energies and compositions of the frontier orbitals for the three molecules (Fig. 1) imply that (while a narrower HOMO–LUMO gap should be observed for the free molecule) BF should be a better σ donor than either N2 or CO (higher HOMO energy and greater localization on the donor atom; Fig. 2) and also a comparable π acceptor (based on a similar LUMO energy but greater localization on the donor atom). | ||
| Fig. 1 HOMO and LUMO energies for the isoelectronic 10 valence electron diatomics N2, CO and BF, plus BNH2 and BO− (adapted from ref. 8c). The percentage character from the more electropositive atom in each orbital is also given. For the purposes of comparison the HOMO and LUMO energies of BF are extrapolated as dashed lines. | ||
 | ||
| Fig. 2 σ Donation and π acceptor behaviour of EX ligands. | ||
Thus, for model complexes such as [(OC)4Fe(EX)] and [(OC)4Co(EX)]+, greater bond dissociation energies are calculated for EX = BF (309 and 296 kJ mol−1, respectively) than for the corresponding carbonyl complexes (203 and 156 kJ mol−1).8 Of relevance to the broader focus of synthetic work (vide infra) are similar computational analyses carried out for the related ligands BNH2 and BO− (Fig. 1). The former species possesses an even smaller HOMO–LUMO gap than BF (suggesting lower kinetic stability) and a non-degenerate set of π type orbitals analogous to the vinylidene (CCR2) ligand family; BO− possesses almost no π acceptor properties due to its exceptionally high energy LUMOs, but very strong σ-donor properties.
Despite the high thermodynamic stabilities calculated for BF and related metal complexes, the highly polar nature of the BF bond and the large build-up of positive charge at boron imply that the kinetic lability of such systems may well be problematic. Shielding of the reactive boron centre via sterically demanding amino groups (BNR2) or by the incorporation of BX in a bridging fashion between two metal centres were suggested as potential solutions; both strategies have now been realised synthetically.7,8 With the latter solution in mind, the increased propensity of BF (over valence isoelectronic group 4 ligands) to adopt bridging (μ2 or μ3) modes of coordination in multimetallic systems has recently been predicted.34,35
The first experimental report of a transition metal complex of BF comes from the early work of Timms, in a brief description of a thermally unstable volatile compound formulated as [(F3P)4Fe(BF)], formed via the condensation of iron vapour with B2F4 and PF3.26 More recently a combination of infrared spectroscopy (Fig. 3) and computation has been used to characterize compounds formulated as [F2M(BF)], among the products of the reactions of metal atoms (M = Ti, Zr, Hf, Th) with BF3/argon at 6 K.36–38 While these tantalising reports hint at the viability of mono-metallic fluoroborylene complexes, structurally authenticated systems containing terminal BF ligands have yet to be reported. Mirroring the predictions of computational studies, the majority of authenticated terminal borylene complexes, [LnM(BX)], instead feature sterically demanding aminoborylene ligands, BNR2 (e.g. R = SiMe3, Cy).7 Moreover, given the lack of readily available sources of ligands of this type (in contrast to CO and N2), a number of alternative synthetic approaches have been developed to give access to such systems; these include double salt elimination,39borane dehydrogenation,40 metal-to-metal borylene transfer,41 and halide abstraction/ejection.42
 | ||
| Fig. 3 Infrared spectra of the products of the reactions of Group 4 metal atoms with BF3/excess argon at 6 K: (a) Ti on deposition; (b) Ti after annealing to 20 K; (c) Ti after irradiation at 220 nm; (d) Zr on deposition; and (e) Hf on deposition. Reproduced with permission from X. Wang, B.O. Roos and L. Andrews, Angew. Chem., Int. Ed., 2010, 49, 157–160 (ref. 36). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. | ||
During the course of these efforts a number of complexes have been reported which feature (terminal) metal-bound B–X fragments, albeit stabilized by coordination at boron of an additional Lewis base. These include the doubly base-stabilized bromoborylene complex [Cp*Fe(CO)2{BBr(4-pic)2}]+Br− (1) formed by halide substitution in [Cp*Fe(CO)2BBr2] in the presence of two equivalents of picoline,43 the related system [Cp′Mn(CO)2{BCl(PdPCy3)2}] containing two boron-bound Pd(PCy3) donors44 and the intriguing dinuclear metallaborane 2, formed by dimerization of the putative terminal chloroborylene [Cp′Mn(CO)2(BCl)], itself generated by the action of CO and UV irradiation on [{Cp′Mn(CO)2]2(μ-BCl)].45

While terminally bound fluoroborylene complexes remain elusive, it is worth noting that very recent work has led to the synthesis of a number of platinum complexes containing the isoelectronic BO− ligand.46–48 Synthetically such systems (e.g. [trans-(Cy3P)2Pt(BO)Br], 3, Scheme 1) have been accessed via initial oxidative addition of one of the B–Br bonds in Br2BOSiMe3 to Pt(0) followed by elimination of BrSiMe3 across the B–O linkage.46 Both the measured BO stretching frequencies for 3 (1853 and 1797 cm−1 for the 10BO and 11BO isotopomers, respectively) and the B–O distance determined for a related thiophenolate derivative [1.210(3) Å] are consistent with the presence of a B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond.473 itself shows impressive thermal stability and resistance to oligomerization; a related cationic system 4, however, formed by halide abstraction, dimerizes to yield a rare example of a B2O2 four-membered ring.47
O triple bond.473 itself shows impressive thermal stability and resistance to oligomerization; a related cationic system 4, however, formed by halide abstraction, dimerizes to yield a rare example of a B2O2 four-membered ring.47
 | ||
| Scheme 1 Synthesis and reactivity of platinum oxyboryl complexes. | ||
A bridging mode of coordination for the BF fragment has proved to be viable, although to date only a single crystallographically characterized example has been reported.38,49 The simple reaction of Na[CpRu(CO)2] with BF3·OEt2, although strongly solvent dependent, leads to the formation of [{CpRu(CO)2}2(μ-BF)] (6) in high yield when diethyl ether is employed as the reaction medium (Scheme 2).49 The structure of 6, featuring an unsupported bridging BF fragment is unprecedented in structurally authenticated carbonyl chemistry. The B–F separation (1.348(3) Å), although markedly shorter than the sum of the relevant covalent radii (1.46 Å) is strongly reminiscent of those found in related difluoroboryl complexes LnM(BF2)x (typically 1.32–1.35 Å).50,51CO can be scavenged from 6 by the use of the electron rich Pt(0) complex [Pt(PCy3)2] to give the spectroscopically characterized (Ru–Ru supported) complex 8, although attempts to use thermolysis, photolysis or amine oxide reagents to bring about the same chemistry were unsuccessful.52 Chemically, the B–F bond itself appears to be prone to heterolytic scission; reaction of 6 with AlCl3 generates the cationic metallaborylene [{CpRu(CO)2}2(μ-B)]+ (7, as the [AlCl4]− salt), in a manner similar to related bridged chloro- and bromoborylene systems, [{Cp′Fe(CO)2}2(μ-BCl)] and [{Mn(CO)5}2(μ-BX)] (X = Cl, Br).53,54 Intriguingly, the related system [{Mn(CO)5}2(μ-B)]+ shows spontaneous reactivity in the reverse sense to generate [Mn(CO)5]2(μ-BF), which is characterized by an 11B NMR shift and 1JBF coupling constant reminiscent of 6 (δB = 123.9 ppm, 1JBF = 265 Hz, cf. 97.3 ppm and 247 Hz for 6). A different chemical approach to B–X bond cleavage is brought about by the reaction of the (methylcyclopentadienyl)manganese chloroborylene system [{Cp′Mn(CO)2}2(μ-BCl)] with lithium powder in dimethoxyethane (DME). Thus, reduction leads to the formation of [Li(DME)3]+[{Cp′Mn(CO)2}2(μ-B)]−, the anionic component of which is isoelectronic to [{Cp′′Fe(CO)2}2(μ-B)]+ and [{CpRu(CO)2}2(μ-B)]+.55
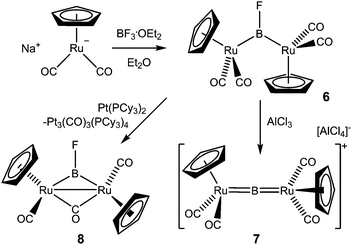 | ||
| Scheme 2 Synthesis and reactivity of a dinuclear ruthenium fluoroborylene complex. | ||
Heavier group 13 element monohalides
The ‘free’ molecules
The monohalides of the Group 13 metals are naturally divided into two groups – those of aluminium and gallium, for which disproportionation into the metal and the metal trihalide is thermodynamically favourable under standard conditions,1 and those of indium and thallium which (with the exception of InF) are stable with respect to disproportionation in the solid state, and which are commercially available.Al(I) halides are entropically favoured in the gas phase at high temperature/low pressure and can be trapped in inert gas matrices. Spectroscopic studies of the AlX diatomics in the gas phase reveal bond lengths of 1.654, 2.130, 2.295 and 2.537 Å for X = F – I, respectively;1,56,57 by contrast the symmetrical halide-bridged dimers, Al(μ-F)2Al and Al(μ-Cl)2Al, predominate in low temperature matrices.58 The related gaseous molecules GaX feature slightly longer bonds than their aluminium counterparts (1.774, 2.202, 2.352 and 2.575 Å, respectively), consistent with a larger covalent radius for Ga(I).1,57 As with aluminium, symmetrically bridged dimers, Ga(μ-X)2Ga, have also been identified spectroscopically for X = F and Cl.4,59 Co-condensation of AlX molecules with mixtures of toluene and donor solvents allows the preparation of metastable solutions of AlX (X = Cl, Br, or I).4–6,60,61 By utilizing NEt3 or PEt3, the Al(I) halide adducts [AlX(EEt3)]4 (E = N, X = Br, I; E = P, X = I) could be isolated and structurally characterized.62 Related Ga(I) species of the type [GaX(L)]n, (X = Cl, Br, I; L = ether, amine, phosphine) have also been prepared,4 and two such adducts, [Ga8I8(PEt3)6] and [Ga10Br10(NC5H4-4-tBu)10], have been crystallographically characterized.61,63
Although no true Ga(I) halides are stable in the solid state, it is worth mentioning in passing reports of a compound initially described in 1955, which was thought to be Ga(I) iodide (GaI), synthesized by heating the respective elements in vacuo.64,65 Subsequent diffraction studies showed that these materials contained the Ga(I)/Ga(II) mixed valence salt [Ga]2[Ga2I6].66 A new synthesis of GaI was reported by Green in 1990, using the ultrasonically activated reaction of gallium metal with iodine in toluene;67 subsequent analysis by Raman spectroscopy, however, revealed that it consisted of a mixture of gallium subhalides, including [Ga]2[Ga2I6].68 Despite this, Green's compound has proved to be a versatile, readily accessible reagent for the synthesis of other Ga(I) species, and to act as a putative source of GaI in insertion reactions with a variety of chemical bonds.67,69,70 This reagent, for example, acts as the ultimate source of the GaI fragment in a recently reported transition metal complex containing the GaI diatomic.71–73
By contrast to aluminium and gallium, the halides InX (X = Cl, Br, I) and TlX (X = F, Cl, Br, I) are stable with respect to disproportionation in the solid state at ambient temperature. This, together with their commercial availability, offers them as viable precursors to low oxidation state indium/thallium compounds, and (in the case of the indium compounds) underpins their increasing use as reagents in organic synthesis.1,3,74,75 In the vapour phase, bond lengths of 1.985 (F), 2.401 (Cl), 2.543 (Br) and 2.754 Å (I) have been determined for the In(I) diatomics, with corresponding distances of 2.084, 2.485, 2.618 and 2.814 Å measured for the corresponding thallium compounds.1,3,57a Moreover, the reactivity of both the singlet (1Σ) and triplet (3Π) states of InCl towards HX (X = H, Cl, or OH) have been studied in argon matrices.59,76,77 As far as solution-phase chemistry is concerned, early reports from Tuck suggested that treatment of In(I) halides with Lewis bases at low temperature could be used to generate soluble complexes which are stable with respect to disproportionation. Thus, solutions of InBr (ca. 16 mM) in TMEDA/toluene mixtures are stable below −20 °C; the crystalline complex InBr(TMEDA) has recently been isolated from a similar solution, showing it to be monomeric, but with long range In⋯In contacts (ca. 3.7 Å) in the solid state.78,79
Coordination chemistry
The coordination chemistry of the heavier Group 13 monohalides at transition metal centres remains an under-developed area, with systems exploiting the steric protection afforded by substituents such as terphenyl, pentamethylcyclopentadienyl, β-diketiminato or guanidinato groups being far more prevalent.7 Thermodynamically, as with boron monohalide ligands, rather high bond dissociation energies have been calculated for such complexes.80 Thus, BDEs that are smaller than those for [Fe(CO)5] (193.7 kJ mol−1 for both axial and equatorial CO ligands) but larger than those of [Fe(CO)4(N2)] (88.7/91.6 kJ mol−1 for axial/equatorial N2) have been calculated for [Fe(CO)4(GaX)] [X = F: 140.6 (axial)/141.8 (equatorial); Cl: 151.5/151.0; Br: 153.6/152.7; I: 158.6/157.7 kJ mol−1]. In addition, while the ratios of covalent to electrostatic interactions in the (CO)4Fe–GaX bonds are very similar to the corresponding values calculated for (CO)4Fe–CO and (CO)4Fe–N2, the importance of σ-donation to the covalent bonding contribution is much stronger than π-back-donation for GaX [while being of similar magnitude for both (CO)4Fe–CO and (CO)4Fe–N2 bonds].80Complexes containing the heavier Group 13 monohalides as terminally bound ligands have only recently been realised experimentally (vide infra),71–73 although a number of systems have previously been reported which feature these EX fragments bridging between two metal centres. These include systems featuring sterically less encumbered transition metal fragments which adopt dimeric structures of the type [(LnM)2E(μ-X)2E(MLn)2] or related polymeric architectures.70,81–85 Mononuclear systems can be isolated in such cases by the additional coordination of a Lewis base at the Group 13 centre.83,86–88 By contrast, simple tri-coordinate systems of the type [LnM]2(μ-EX) are surprisingly uncommon,70,89–91 with the tendency of such systems to oligomerize viaE-X-E bridges being modulated by bulkier ancillary ligands, or by the electrostatic repulsion brought about by a net overall charge (as in the cases of [{(OC)5Cr}2(μ-EX)]2−, EX = InBr, TlCl, TlBr, TlI). In the cases of the Cp*Fe(CO)2 systems shown in Scheme 3, synthetic routes originating in E(I) or E(III) precursors are viable (utilizing insertion or salt elimination protocols), with the monomeric complexes obtained contrasting with the oligo/polymeric structures of the related CpFe(CO)2 compounds.70,83,92,93 Mechanistically, there are clear analogies between the reaction of [Cp*Fe(CO)2]2 and InI (to yield 9-I) and classical oxidative insertion reactions; that said, the very similar electronegativities of iron and indium (1.83 and 1.78, respectively, on the Pauling scale) mean that the assignments of formal oxidation states in the product are somewhat arbitrary. In similar fashion, the reaction of [Cp*Fe(CO)2]− with the metal(III) halides GaCl3 and InBr3 might be regarded as simple halide/metal anion metathesis, although here too formal oxidation states are ill-defined (Pauling electronegativity for gallium = 1.81).
 | ||
| Scheme 3 Syntheses of monomeric bridged EX complexes from E(I) and E(III) precursors. | ||
The first examples of transition metal complexes featuring terminally bound EX fragments were a range of base-stabilized complexes reported by Fischer and co-workers in 1998.94,95 Synthesized by the reactions of carbonylate dianions {e.g.[Fe(CO)4]2−, [Cr(CO)5]2− and [W(CO)5]2−} with Group 13 trihalides in the presence of a chelating Lewis base, complexes such as 12 and 13 feature strongly σ-donating (but weakly π accepting) E(X)L2 ligands. The metal–ligand bonds in these systems are relatively long {e.g. 2.337(1) Å for 12cf. 2.225(1) Å for [(OC)4FeGa(C6H3-Trip2-2,6)]},96 and the E–X distances are also reflective of single bond character [e.g. d(Ga–I) = 2.642(1) Å for 12, cf. 2.701(1) Å for 9 and ca. 2.63 Å for the sum of the respective covalent radii].70,94,97 In related chemistry, the cationic derivative [Cp*Fe(CO)2{GaCl(phen)}]+ (14) has been synthesized as the [BPh4]− salt via the reaction of [Cp*Fe(CO)2GaCl2] with the halide abstraction agent Na[BPh4] in the presence of 1,10-phenanthroline (phen).98

Halide abstraction chemistry can also be used to access two-coordinate group 13 ligand systems. This methodology was initially applied in 2004 in the synthesis of the trimetallic indium and gallium cations 15 and 16, and subsequently adapted for the synthesis of the iodogallylene complex [Cp*Fe(dppe)(GaI)]+ [BArf4]−, 17, by employing a more electron-rich and sterically encumbered bis(phosphine)iron fragment.71–73,89,90 This approach circumvents the synthetic obstacle presented by the lack of tractable solution-phase sources of the EX molecule, and complex 17 represents the first example of terminal coordination of such a diatomic. Relevant geometric parameters determined crystallographically [viz. d(Fe–Ga) = 2.222(1) Å, d(Ga–I) = 2.444(1) Å, ∠(Fe–Ga–I) = 171.4(1)°] are consistent with the low coordination number at gallium, cf. d(Fe–Ga) = 2.225(1) Å for the similarly two-coordinate ligand system in [(OC)4FeGa(C6H3-Trip2-2,6)], but 2.305(1) Å for (tetra-coordinate) 14.71,96 The Ga–I separation [2.444(1) Å] is similarly short [cf. 2.630 Å (mean) for the diiodogallyl precursor and 2.575 Å for the gas-phase GaI diatomic].71,57 While quantum chemical analyses for the cationic component of 17 reveal molecular orbitals consistent with σ-donor and π-acceptor behaviour for the GaI ligand, the overall covalent (orbital) contribution to the M-GaI bond is calculated to be relatively low (−236 kJ mol−1cf. analogous values of −469, −397 and −262 kJ mol−1 for the related complexes of BF, CO and N2) and indeed is found to be comparable to the electrostatic binding contribution (−234 kJ mol−1). Presumably, despite the higher energy of the HOMO for GaI (−6.08 eV cf. −9.03 eV for CO) and the greater localization of the LUMO at the donor atom, the weaker orbital contribution for GaI reflects the more diffuse nature of the 4s/4p derived orbitals at gallium and less effective interaction with the fragment orbitals of [CpFe(dmpe)]+.
Overall metal–ligand bond strengths [ΔEint = −103 (GaI), −285 (BF), −213 (CO) and −120 kJ mol−1 (N2)] reveal relatively weak binding of the GaI ligand, which (consistent with this assertion) can be displaced quantitatively from 17 by the addition of CO to give [Cp*Fe(dppe)(CO)]+[BArf4]− (Scheme 4). In the absence of such reagents, 17 is stable for weeks in fluorobenzene solution, primarily reflecting (i) effective steric shielding of the gallium centre by the ancillary phosphine and Cp* ligands; and (ii) a net cationic charge which retards the tendency towards dimerization found in putative charge-neutral systems such as [Cp′Mn(CO)2(BCl)].45
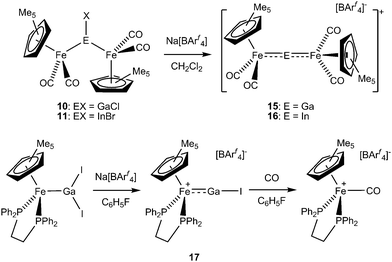 | ||
| Scheme 4 Cationic complexes featuring two-coordinate gallium/indium containing ligands formed by halide abstraction. | ||
Although 17 retains a unique place as a valence isoelectronic Group 13/Group 17 analogue of CO and N2, a related complex of interest is the similarly sterically unencumbered methylgallylene complex [(Cp*Ga)4Rh(GaMe)]+[BArf4]−, synthesized by protonolysis of the methyl(η1-pentamethylcyclopentadienyl)gallyl precursor [(Cp*Ga)4Rh{Ga(η1-Cp*)Me}].99 Interestingly, the Rh–GaMe distance [2.471(1) Å] becomes noticeably shorter [2.334(1) Å] on coordination of pyridine at the gallium centre; such behaviour contrasts markedly with related borylene systems,100 and presumably reflects not only the gallylene ligand becoming a better σ-donor, but also the relative unimportance of π back-bonding in this system.
Conclusions
While the Group 13 monohalides have a history spanning more than 70 years, and their use in the synthesis of low-valent alkyl, aryl, amido and related derivatives has progressed rapidly in recent years, particularly through the work of Power, Schnöckel, Fischer and Jones, the coordinative trapping of the simple diatomic molecules, EX, has only recently been achieved. In part of course, this reflects the fact that (unlike valence isoelectronic ligands such as CO or N2) sources of EX (E = B, Al, Ga) are known, as donor-free species, only under conditions of extreme temperature, with problems stemming from disproportionation or aggregation inherent at (or close to) room temperature. However, the recent structural characterization of complexes containing bridging BF and terminal GaI fragments (i.e. [{CpRu(CO)2}2(μ-BF)], 6,49 and [Cp*Fe(dppe)(GaI)]+ [BArf4]−, 17)71–73 allow questions concerning the geometric/electronic structure of such systems to be tackled. Concerning the dinuclear complex 6, a point of interest arises as to whether the BF ligand binds to the two metal centres in the manner characteristic of μ2–CO ligands (i.e. with the ligand formally being in a singlet state),101,102 or whether the boron centre is effectively trigonal, and hence the BF fragment formally derived from a triplet state. The latter situation would, of course be analogous, for example, to the bonding of the CO unit in a ketone and related to the [{CpMn(CO)}2(μ-BtBu)] system investigated by Stalke and Braunschweig.103 Structural data, and in particular the wide Ru–B–Ru angle found for [{CpRu(CO)2}2(μ-BF)] [131.4(1)°],49 imply that the latter description may have some validity, i.e. compound 6 might best be described formally in terms of the interaction between a triplet BF fragment and two [CpRu(CO)2] fragments (cf. a dimetallaborane; Fig. 4). From an energetic perspective the singlet–triplet gap for BF has been calculated to be ca. 86 kcal mol−1 (with a singlet ground state),104 but with M–B bonds in related systems worth (in energetic terms) in the order of 60–70 kcal mol−1 [e.g. 66 kcal mol−1 for the Fe–B bond in CpFe(CO)2BF2],105 the hypothetical formation of 6 from triplet BF and two [CpRu(CO)2]· fragments is therefore feasible.106![Interaction between [CpRu(CO)2] and triplet BF fragments.](/image/article/2011/SC/c0sc00508h/c0sc00508h-f4.gif) | ||
| Fig. 4 Interaction between [CpRu(CO)2] and triplet BF fragments. | ||
Such a description is consistent with the BF bond length for 6 [1.348(3) Å]49 which although somewhat shorter than the sum of the covalent radii for boron and fluorine (1.46 Å),97 is significantly longer than the formal triple bond in the BF diatomic (1.263 Å).22 By contrast, the cationic terminal iodogallylene complex 17 features a GaI distance, which at 2.444(1) Å, is shorter even than that in the parent diatomic (2.575 Å). The Fe–Ga distance is also found to be short, even with due allowance made for the low coordination number at gallium. This observation is attributed (in the main) not to significant M–Ga π orbital interactions, but to high gallium s-orbital contributions to the M–Ga bonding orbitals.107 Consistent with this assertion, ΔEσ is found to be overwhelmingly the dominant contribution to the orbital interaction between model [CpFe(PMe3)2]+ and [GaI] fragments (with ΔEπ equating to only 18% of the total orbital contribution); the GaI ligand thus behaves predominantly as a σ-donor. Interestingly, quantum chemical studies of related charge neutral complexes containing the GaI ligand, notably [axial-Fe(CO)4(GaI)], reveal remarkably similar geometric/electronic properties to those of the cationic system 17, viz. very short Fe–Ga and GaI distances, similar ratios of electrostatic to covalent bonding contributions to the total interaction energy (ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and a similarly small role for π symmetry orbital interactions (ca. 20%). Such findings hint at a similar mode of attachment for the GaI ligand in these two types of system.80,107
:1), and a similarly small role for π symmetry orbital interactions (ca. 20%). Such findings hint at a similar mode of attachment for the GaI ligand in these two types of system.80,107
Given the long established track-record in coordination chemistry of the trapping and subsequent spectroscopic/structural interogation of otherwise kinetically very labile species, and the recent isolation of complexes containing terminally bound BO−, CF, GaI and even Ga+ ligands,46–48,71–73,108,109 it seems likely that further advances in the field will soon be forthcoming. An attractive target would be a terminally bound complex of BF, offering as it would, precise experimental comparison of electronic structure with the textbook systems CO and N2. While quantum chemical studies offer encouragement as to the thermodynamic stability of such complexes, it is the design of innovative preparative-scale routes to suitably kinetically frustrated species that once again holds the experimental key. Comparison of experimental data with theory would then offer to inform the ongoing debate about potential bonding mode(s) in these compounds.
Notes and references
- (a) A. J. Downs, Chemistry of Aluminium, Gallium, Indium and Thallium; Blackie, Glasgow, 1993 Search PubMed; (b) S. Aldridge and A. J. Downs, The Chemistry of the Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities; Wiley, Chichester, 2011 Search PubMed.
- For a recent review of frustrated Lewis pairs, see: D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 56–76 Search PubMed.
- J. A. J. Pardoe and A. J. Downs, Chem. Rev., 2007, 107, 2–45 CrossRef CAS.
- C. Dohmeier, D. Loos and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129–149 CrossRef CAS.
- A. Schnepf and H. Schnöckel, Angew. Chem., Int. Ed., 2002, 41, 3532–3554 CrossRef CAS.
- K. Koch, R. Burgert and H. Schnöckel, Angew. Chem., Int. Ed., 2007, 46, 5795–5798 CrossRef CAS.
- (a) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS; (b) H. Braunschweig, R. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS; (c) D. Vidovic, G. A. Pierce and S. Aldridge, Chem. Commun., 2009, 1157–1171 RSC; (d) E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS; (e) R. J. Baker and C. Jones, Coord. Chem. Rev., 2005, 249, 1857–1869 CrossRef; (f) C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 4161–4176 CrossRef CAS; (g) R. A. Fischer and J. Weiss, Angew. Chem., Int. Ed., 1999, 38, 2831–2850 CAS.
- (a) F. M. Bickelhaupt, U. Radius, A. W. Ehlers, R. Hoffmann and E. J. Baerends, New J. Chem., 1998, 22, 1–3 RSC; (b) U. Radius, F. M. Bickelhaupt, A. W. Ehlers, N. Goldberg and R. Hoffmann, Inorg. Chem., 1998, 37, 1080–1090 CrossRef CAS; (c) A. W. Ehlers, E. J. Baerends, F. M. Bickelhaupt and U. Radius, Chem.–Eur. J., 1998, 4, 210–221 CrossRef CAS.
- R. L. Altman, J. Chem. Phys., 1959, 31, 1035–1038 CrossRef CAS.
- R. F. Barrow, Trans. Faraday Soc., 1960, 56, 952–958 RSC.
- D. W. Robinson, J. Mol. Spectrosc., 1963, 11, 275–300 CrossRef CAS.
- R. K. Nesbet, J. Chem. Phys., 1964, 40, 3619–3633 CrossRef CAS.
- J. Blauer, M. A. Greenbaum and M. Farber, J. Phys. Chem., 1964, 68, 2332–2334 CrossRef CAS.
- W. M. Huo, J. Chem. Phys., 1965, 43, 624–647 CAS.
- D. L. Hildenbrand and E. Murad, J. Chem. Phys., 1965, 43, 1400–1403 CrossRef CAS.
- R. B. Caton and A. E. Douglas, Can. J. Phys., 1970, 48, 432–452 CAS.
- J. Singh, K. P. R. Nair and D. K. Rai, J. Mol. Struct., 1970, 6, 328–332 CrossRef CAS.
- F. J. Lovas and D. R. Johnson, J. Chem. Phys., 1971, 55, 41–44 CrossRef CAS.
- V. S. Kushawaha, B. P. Asthana and C. M. Pathak, Spectrosc. Lett., 1972, 5, 357–360 CrossRef CAS.
- J. M. Dyke, C. Kirby and A. Morris, J. Chem. Soc., Faraday Trans. 2, 1983, 79, 483–490 RSC.
- H. Bredohl, I. Dubois, F. Melen and M. Vervloet, J. Mol. Spectrosc., 1988, 129, 145–150.
- G. Cazzoli, L. Cludi, C. Degli Esposti and L. Dore, J. Mol. Spectrosc., 1989, 134, 159–167 CrossRef CAS.
- K. Q. Zhang, B. Guo, V. Braun, M. Dulick and P. F. Bernath, J. Mol. Spectrosc., 1995, 170, 82–93 CrossRef CAS.
- P. L. Timms, J. Am. Chem. Soc., 1967, 89, 1629–1623 CrossRef CAS.
- P. L. Timms, J. Am. Chem. Soc., 1968, 90, 4585–4589 CrossRef CAS.
- P. L. Timms, Acc. Chem. Res., 1973, 6, 118–123 CrossRef CAS.
- (a) T. Mennekes, P. Paetzold, R. Boese and D. Blaser, Angew. Chem., Int. Ed. Engl., 1991, 30, 173–175 CrossRef; (b) C.-J. Maier, H. Pritzkow and W. Siebert, Angew. Chem., Int. Ed., 1999, 38, 1666–1668 CrossRef CAS; (c) H. Binder, R. Kellner, K. Vaas, M. Hein, F. Baumann, M. Wanner, R. Winter, W. Kaim, W. Hönle, Y. Grin, U. Wedig, M. Schultheiss, R. K. Kremer, H. G. von Schnering, O. Groeger and G. Engelhardt, Z. Anorg. Allg. Chem., 1999, 625, 1059–1072 CrossRef CAS.
- J. A. J. Pardoe, N. C. Norman, P. L. Timms, S. Parsons, I. Mackie, C. R. Pulham and D. W. H. Rankin, Angew. Chem., Int. Ed., 2003, 42, 571–573 CrossRef CAS.
- P. L. Timms, N. C. Norman, J. A. J. Pardoe, I. D. Mackie, S. L. Hinchley, S. Parsons and D. W. H. Rankin, Dalton Trans., 2005, 607–616 RSC.
- J. A. J. Pardoe, N. C. Norman and P. L. Timms, Polyhedron, 2002, 21, 543–548 CrossRef.
- P. Hassanzadeh and L. Andrews, J. Phys. Chem., 1993, 97, 4910–4915 CrossRef CAS.
- J. C. Jeffery, N. C. Norman, J. A. J. Pardoe and P. L. Timms, Chem. Commun., 2000, 2367–2368 RSC.
- B. Pachaly and R. West, Angew. Chem., Int. Ed. Engl., 1984, 23, 454–455 CrossRef.
- L. Xu, Q. Li, Y. Xie, R. B. King and H. F. Schaefer III, Inorg. Chem., 2010, 49, 2996–3001 CrossRef CAS.
- L. Xu, Q. Li, Y. Xie, R. B. King and H. F. Schaefer III, Inorg. Chem., 2010, 49, 1046–1055 CrossRef CAS.
- X. Wang, B. O. Roos and L. Andrews, Angew. Chem., Int. Ed., 2010, 49, 157–160 CAS.
- X. Wang, B. O. Roos and L. Andrews, Chem. Commun., 2010, 46, 1646–1648 RSC.
- H. Braunschweig and R. Dewhurst, Angew. Chem., Int. Ed., 2010, 49, 3412–3414 CrossRef CAS (highlight).
- (a) A. H. Cowley, V. Lomelí and A. Voigt, J. Am. Chem. Soc., 1998, 120, 6401–6402 CrossRef CAS; (b) H. Braunschweig, C. Kollann and U. Englert, Angew. Chem., Int. Ed., 1998, 37, 3179–3180 CrossRef CAS.
- G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878–12879 CrossRef CAS.
- H. Braunschweig, M. Colling, C. Kollann, H. G. Stammler and B. Neumann, Angew. Chem., Int. Ed., 2001, 40, 2298–2300 CrossRef CAS.
- (a) D. L. Coombs, S. Aldridge, C. Jones and D. J. Willock, J. Am. Chem. Soc., 2003, 125, 6356–6357 CrossRef CAS; (b) D. Vidovic, M. Findlater, G. Reeske and A. H. Cowley, Chem. Commun., 2006, 3786–3787 RSC; (c) D. A. Addy, G. A. Pierce, D. Vidovic, D. Mallick, E. D. Jemmis, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 4586–4588 CrossRef CAS.
- H. Braunschweig, K. Radacki, F. Seeler and G. R. Whittell, Organometallics, 2006, 25, 4605–4610 CrossRef CAS.
- H. Braunschweig, M. Burzler, R. D. Dewhurst, K. Radacki and Fabian Seeler, Z. Anorg. Allg. Chem., 2008, 634, 1875–1879 CrossRef CAS.
- H. Braunschweig, M. Colling, C. Hu and K. Radacki, Angew. Chem., Int. Ed., 2002, 41, 1359–1361 CrossRef CAS.
- (a) H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345–347 CrossRef CAS; (b) S. Westcott, Angew. Chem., Int. Ed., 2010, 49 DOI:10.1002/anie.201003379 , in press, (highlight).
- H. Braunschweig, K. Radacki and A. Schneider, Angew. Chem., Int. Ed, 2010, 49, 5993–5996 CAS.
- H. Braunschweig, K. Radacki and A. Schneider, Chem. Commun., 2010, 46, 6473–6475 RSC.
- D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed., 2009, 48, 3669–3672 CrossRef CAS.
- Difluoroboryl complexes: see ref. 43 and (a) A. Kerr, T. B. Marder, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, P. L. Timms and G. R. Whittell, Chem. Commun., 1998, 319–320 RSC; (b) N. Lu, N. C. Norman, A. G. Orpen, M. J. Quayle, P. L. Timms and G. R. Whittell, J. Chem. Soc., Dalton Trans., 2000, 4032–4037 RSC.
- Mono-fluoroboryl complexes: (a) Z. Lu, C.-H. Jun, S. R. de Gala, M. Sigalas, O. Eisenstein and R. H. Crabtree, J. Chem. Soc., Chem. Commun., 1993, 1877–1880 RSC; (b) Z. Lu, C.-H. Jun, S. R. de Gala, M. P. Sigalas, O. Eisenstein and R. H. Crabtree, Organometallics, 1995, 14, 1168–1175 CrossRef CAS; (c) H. Braunschweig, M. Colling, C. Kollann and U. Englert, J. Chem. Soc., Dalton Trans., 2002, 2289–2296 RSC; (d) D. L. Coombs, S. Aldridge, A. Rossin, C. Jones and D. J. Willock, Organometallics, 2004, 23, 2911–2926 CrossRef CAS.
- D. Vidovic and S. Aldridge, unpublished results.
- P. Bissinger, H. Braunschweig and F. Seeler, Organometallics, 2007, 26, 4700–4701 CrossRef CAS.
- H. Braunschweig, K. Kraft, T. Kupfer, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2008, 47, 4931–4933 CrossRef CAS.
- H. Braunschweig, M. Burzler, R. D. Dewhurst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5650–5653 CrossRef CAS.
- (a) W. Klemm and E. Voss, Z. Anorg. Allg. Chem., 1943, 251, 233–240 CrossRef CAS.
- (a) K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules, van Nostrand Reinhold, New York, 1979 Search PubMed; (b) H. G. Hedderich, M. Dulick and P. F. Bernath, J. Chem. Phys., 1993, 99, 8363–8370 CrossRef CAS.
- (a) R. Ahlrichs, L. Zhenyan and H. Schnöckel, Z. Anorg. Allg. Chem., 1984, 519, 155–164 CAS; (b) H.-J. Himmel, Eur. J. Inorg. Chem., 2005, 1886–1894 CrossRef CAS.
- H.-J. Himmel, Dalton Trans., 2003, 3639–3649 RSC.
- M. Tacke and H. Schnöckel, Inorg. Chem., 1989, 28, 2895–2896 CrossRef CAS.
- C. U. Doriat, M. Friesen, E. Baum, A. Ecker and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1997, 36, 1969–1971 CrossRef CAS.
- (a) M. Mocker, C. Robl and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1994, 33, 1754–1755 CrossRef; (b) A. Ecker and H. Schnöckel, Z. Anorg. Allg. Chem., 1996, 622, 149–152 CrossRef CAS; (c) A. Ecker and H. Schnöckel, Z. Anorg. Allg. Chem., 1998, 624, 813–816 CrossRef CAS; (d) A. Ecker, R. Köppe, C. Üffing and H. Schnöckel, Z. Anorg. Allg. Chem., 1998, 624, 817–822 CrossRef CAS.
- T. Duan, G. Stösser and H. Schnöckel, Angew. Chem., Int. Ed., 2005, 44, 2973–2975 CrossRef CAS.
- J. D. Corbett and R. K. McMullan, J. Am. Chem. Soc., 1955, 77, 4217–4219 CrossRef CAS.
- M. Wilkinson and I. J. Worrall, J. Organomet. Chem., 1975, 93, 39–42 CrossRef CAS.
- C. Gerlach, W. Hönle and A. Simon, Z. Anorg. Allg. Chem., 1982, 486, 7–21 CrossRef CAS.
- M. L. H. Green, P. Mountford, G. J. Smout and S. R. Speel, Polyhedron, 1990, 9, 2763–2765 CrossRef CAS.
- S. Coban, Diplomarbeit, Universität Karlsruhe, 1999 Search PubMed.
- R. J. Baker and C. Jones, Dalton Trans., 2005, 1341–1348 RSC.
- N. R. Bunn, S. Aldridge, D. L. Kays (née Coombs), N. D. Coombs, J. K. Day, L.-L. Ooi, S. J. Coles and M. B. Hursthouse, Organometallics, 2005, 24, 5879–5890 CrossRef CAS.
- N. D. Coombs, W. Clegg, A. L. Thompson, D. J. Willock and S. Aldridge, J. Am. Chem. Soc., 2008, 130, 5449–5451 CrossRef CAS.
- N. D. Coombs, D. Vidovic, J. K. Day, A. L. Thompson, D. D. Le Pevelen, A. Stasch, W. Clegg, L. Russo, L. Male, M. B. Hursthouse, D. J. Willock and S. Aldridge, J. Am. Chem. Soc., 2008, 130, 16111–16124 CrossRef CAS.
- H.-J. Himmel and G. Linti, Angew. Chem., Int. Ed., 2008, 47, 6326–6328 CrossRef CAS (highlight).
- (a) H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1985, 24, 893–904 CrossRef; (b) H. Schmidbaur and A. Schier, Organometallics, 2008, 27, 2361–2395 CrossRef CAS.
- D. G. Tuck, Chem. Soc. Rev., 1993, 22, 269–276 RSC.
- H.-J. Himmel, A. J. Downs and T. M. Greene, J. Am. Chem. Soc., 2000, 122, 922–930 CrossRef CAS.
- H.-J. Himmel, J. Chem. Soc., Dalton Trans., 2002, 2678–2682 RSC.
- C. Peppe, D. G. Tuck and L. Victoriano, J. Chem. Soc., Dalton Trans., 1982, 2165–2168 RSC.
- S. P. Green, C. Jones and A. Stasch, Chem. Commun., 2008, 6285–6287 RSC.
- J. A. Gámez, R. Tonner and G. Frenking, Organometallics, 2010, 29, 5676–5680 CrossRef CAS.
- H.-J. Haupt, W. Wolfes and H. Preut, Inorg. Chem., 1976, 15, 2920–2927 CrossRef CAS.
- H.-J. Haupt, H. Preut and W. Wolfes, Z. Anorg. Allg. Chem., 1979, 448, 93–99 CAS.
- L. M. Clarkson, N. C. Norman and L. J. Farrugia, Organometallics, 1991, 10, 1286–1292 CrossRef CAS.
- J. C. Calabrese, L. M. Clarkson, T. B. Marder, N. C. Norman and N. J. Taylor, J. Chem. Soc., Dalton Trans., 1992, 3525–3529 RSC.
- J. J. Schneider, U. Denninger, J. Hagen, C. Krüger, D. Bläser and R. Boese, Chem. Ber., 1997, 130, 1433–1440 CrossRef CAS.
- L. M. Clarkson, W. Clegg, D. C. R. Hockless, N. C. Norman, L. J. Farrugia, S. G. Bott and J. L. Atwood, J. Chem. Soc., Dalton Trans., 1991, 2241–2252 RSC.
- G. Linti, G. Li and H. Pritzkow, J. Organomet. Chem., 2001, 626, 82–91 CrossRef CAS.
- H. Nakazawa, M. Itazaki and M. Owaribe, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m945–m946 CrossRef.
- N. R. Bunn, S. Aldridge, D. L. Kays, N. D. Coombs, A. Rossin, D. J. Willock, J. K. Day, C. Jones and L.-L. Ooi, Organometallics, 2005, 24, 5891–5900 CrossRef CAS.
- N. R. Bunn, S. Aldridge, D. L. Coombs, A. Rossin, D. J. Willock, C. Jones and L.-L. Ooi, Chem. Commun., 2004, 1732–1733 RSC.
- O. J. Curnow, B. Schiemenz, G. Huttner and L. Zsolnai, J. Organomet. Chem., 1993, 459, 17–20 CrossRef CAS.
- A. S. Borovik, S. G. Bott and A. R. Barron, Organometallics, 1999, 18, 2668–2676 CrossRef CAS.
- N. D. Coombs, J. K. Day, S. Aldridge, S. J. Coles and M. B. Hursthouse, Main Group Met. Chem., 2007, 30, 195–198 CAS.
- R. A. Fischer, M. M. Schulte, J. Weiss, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking, C. Boehme and S. F. Vyboishchikov, J. Am. Chem. Soc., 1998, 120, 1237–1248 CrossRef CAS.
- H. Fölsing, O. Segnitz, U. Bossek, K. Merz, M. Winter and R. A. Fischer, J. Organomet. Chem., 2000, 606, 132–140 CrossRef CAS.
- J. Su, X.-W. Li, R. C. Crittendon, C. F. Campana and G. H. Robinson, Organometallics, 1997, 16, 4511–4513 CrossRef CAS.
- J. Emsley, The Elements, OUP, Oxford, 1998 Search PubMed.
- K. Ueno, T. Watanabe and H. Ogino, Appl. Organomet. Chem., 2003, 17, 403 CrossRef CAS.
- T. Cadenbach, C. Gemel, D. Zacher and R. A. Fischer, Angew. Chem., Int. Ed., 2008, 47, 3438–3441 CrossRef CAS.
- S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D. L. Kays (née Coombs), N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118–6122 CrossRef CAS.
- E. D. Jemmis, A. R. Pinhas and R. Hoffmann, J. Am. Chem. Soc., 1980, 102, 2576–2585 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry; University Science Books, Sausalito, California, USA, 2010, pp 29-30 Search PubMed.
- U. Flierler, M. Burzler, D. Leusser, J. Henn, H. Ott, H. Braunschweig and D. Stalke, Angew. Chem., Int. Ed., 2008, 47, 4321–4325 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1999, 103, 8861–8869 CrossRef CAS.
- A. Dickinson, D. J. Willock, R. J. Calder and S. Aldridge, Organometallics, 2002, 21, 1146–1157 CrossRef CAS.
- A further point concerning complexes of the type [(LnM)2(μ-BX)] revolves around the assignment of formal oxidation states. Thus, for complexes of the type [{(η5-C5R5)M(CO)2}2(μ-BX)] (M = Fe, Ru) for example, textbook electronegativity data (Pauling: B 2.04, Fe 1.83, Ru 2.2; absolute scale: B 4.29 eV, Fe 4.06 eV, Ru 4.5 eV)97 imply different metal (and hence boron) oxidation states for the iron and ruthenium complexes, despite their similar spectroscopic data (e.g.CO stretches of 2012, 1960 for 6; 2015, 1935 for [{CpFe(CO)2}2(μ-BCl)]). Thus, it is clear that apportioning oxidation states in this manner between two elements of comparable electronegativity is somewhat arbitrary.
- K. K. Pandey and S. Aldridge, Inorg. Chem., 2011, 50 Search PubMed , asap (10.1021/ic102217z).
- H. Huang, R. P. Hughes, C. R. Landis and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 7454–7455 CrossRef CAS.
- (a) B. Buchin, C. Gemel, T. Cadenbach, R. Schmid and R. A. Fischer, Angew. Chem., Int. Ed., 2006, 45, 1674–1674 CrossRef; (b) S. Aldridge, Angew. Chem., Int. Ed., 2006, 45, 8097–8099 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |