Proximity-driven metallopeptide catalysis: Remarkable side-chain scope enables modification of the Fos bZip domain†
Brian V.
Popp
and
Zachary T.
Ball
*
Department of Chemistry, Rice University, MS 60, 6100 Main Street, Houston, TX 77004. E-mail: zb1@rice.edu; Fax: +1-713-348-5155; Tel: +1-713-348-6159
First published on 21st January 2011
Abstract
Coiled-coil assembly of substrate peptides with dirhodium metallopeptide catalysts enables side-chain modification on the basis of molecular shape. A wide range of amino acids are effectively modified, including the first examples of carboxamide (glutamine and asparagine) modification. The method is used to achieve covalent modification of the c-Fos bZip domain at different residues, depending on the metallopeptide structure. By combining promiscuous catalytic reactivity with specific molecular recognition, this work establishes a general strategy for protein modification on the basis of molecular shape. A broad range of peptide–protein interactions are potentially amenable to this approach.
Introduction
Post-translational covalent modification of proteins is a demanding challenge, requiring bond formation at specific residues in a functional-group rich environment. While living systems routinely accomplish this task,1 manipulating enzymatic function is difficult,2–7 and existing chemical approaches are limited.8,9 In this article, we describe a general strategy for protein modification by combining molecular recognition with a transition-metal catalyst capable of promiscuous side-chain modification, enabling site-specific modification on the basis of molecular shape rather than functional-group reactivity. After developing this concept with coiled-coil models, we describe selective modification of a natural bZip domain.Chemical approaches to protein modification are dominated by residue-specific reactivity, such as cysteine and lysine bioconjugation with electrophilic reagents10,11 or tyrosine alkylation.12–15 However, the scope of side chains accessible to known methods is limited, and a targeted residue is rarely unique within a protein sequence. Because proteins typically contain many copies of a given amino acid, modification of a specific residue requires recognition of local sequence or structure. However, chemical reactions that override inherent reactivity are rare. For polyfunctional metabolite substrates, an approach toward selective reactivity has been developed16,17 based on peptide catalyst screening.18–20 For peptides and proteins, site-specific modification can be achieved in special cases, most notably enzymes, or through recombinant technology to introduce targetable sequences.21 Examples of these approaches include reactive enzyme inhibitors,1,22 photoaffinity reagents,23 templated amide–bond formation,24–26 and reagents that recognize specific repetitive sequences.27–29 However, a general approach for the modification of natural proteins, especially of non-enzymes, would be a valuable new tool.
We have reported30 that supramolecular assembly enables dirhodium metallopeptides to catalyze side-chain modification with significant rate enhancements (≥103) and expands the scope of target residues from tryptophan31 to the other aromatic residues, phenylalanine and tyrosine (Fig. 1). We now disclose that dirhodium metallopeptides are capable of modifying over half of the natural amino-acid side chains, including several residues for which no other modification methods exist, and that our approach enables the covalent modification of a natural coiled-coil domain on the basis of weak, transient supramolecular interactions.
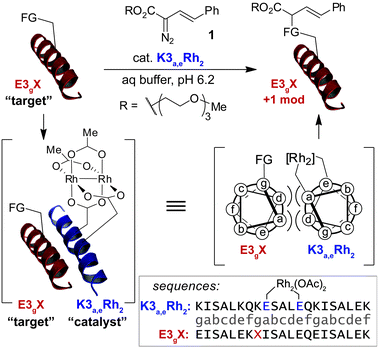 | ||
| Fig. 1 Catalytic covalent modification of amino-acid side chains is achieved using coiled-coil assembly to drive localization of a dirhodium metallopeptide to a specific side chain of a substrate peptide. Label subscripts represent the position, on a helical-wheel model, of key residues that deviate from the parent E3 or K3 sequences. | ||
Results and discussion
Proximity-accelerated side-chain modifcation
To examine the amino-acid scope of proximity-driven dirhodium catalysis, we prepared peptide substrates designed to interact with a metallopeptide catalyst through coiled-coil assembly (Fig. 1). Our design, based on the E3/K3 assembly,32–34 positions a dirhodium center on metallopeptide K3a,eRh2 near position g of the complementary peptide E3gX.35,36 We then varied the residue at position g and examined covalent modification.Remarkably, over half of the natural amino acids are substrates for covalent modification with the styryl-diazo reagent 1 (Table 1). In addition to the previously reported aromatic residues9 (entries 1–3), cysteine, glutamine, asparagine, arginine, and glutamic acid are each modified in >65% conversion within 24 h upon treatment with the metallopeptide catalyst, K3a,eRh2, and diazo 1 (entries 4–7, 9).37 For several other residues—aspartatic acid, serine, histidine, and lysine—modification occurs but more modest conversions are observed (entries 8, 10–12). Relative modification rates (krel) were evaluated based on conversion at 1 h, correcting for catalyst loading and assuming first-order catalyst behavior. The aromatic side chains (entries 1–3, 11) span an especially large range of reactivity. No evidence of modification was observed for threonine, methionine, or any hydrocarbon side chain (entries 13–16). For a few of the newly discovered reactive side chains, a small amount of double modification is observed (entries 4,7–9).
| Entry | E3gX X = | Mol % K3a,eRh2 | Proposed product bond connectivityb | % Conv at 24 hc | k rel |
|---|---|---|---|---|---|
| a All reactions were 50 μM in substrate and 2.5 mM in diazo 1 in aqueous buffer (pH 6.2) at room temperature, unless otherwise noted. b Illustrated structures are proposed based on small-molecule precedent. See text for discussion and Chart S1 for a description of all isomeric possibilities. c Conversion based on MALDI–TOF MS analysis of the reaction mixture. Numbers in parenthesis indicate the ratio of singly modified to doubly modified peptide, in cases where doubly modified species were observed. d Based on initial rate measurements of product formation at 1 h and corrected for catalyst concentration relative to tyrosine modification rate = 100. e A small amount (<10%) of triply modified peptide was observed. f Conversion at 6 h. g 5 mM diazo 1 added in two portions. h Other isomers are possible. See text and eqn (1). | |||||
| 1 | Trp | 1 |
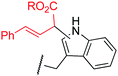
|
> 95 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3)e,f :1.3)e,f |
3000 |
| 2 | Tyr | 10 |
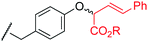
|
> 95 (7:1)g |
100 |
| 3 | Phe | 10 |
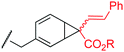
|
> 95 (8:1)g |
87 |
| 4 | Cys | 10 |
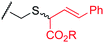
|
78 (5:1) |
560 |
| 5 | Gln | 10 |
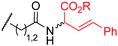
|
> 95f | 280 |
| 6 | Asn | 10 | 70 | 110 | |
| 7 | Glu | 20 |
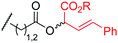
|
76 (9:1)g |
130 |
| 8 | Asp | 20 | 53 (5:1)g |
100 | |
| 9 | Arg | 50 |
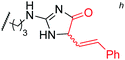
|
73 (3:1)e,g |
97 |
| 10 | Ser | 50 |
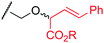
|
28g | 32 |
| 11 | His | 50 |
|
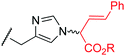
17g | 18 |
| 12 | Lys | 50 |
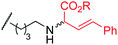
|
8g | 11 |
| 13 | Thr | 50 | –– | NR | –– |
| 14 | Val | 50 | –– | NR | –– |
| 15 | Ala | 50 | –– | NR | –– |
| 16 | Met | 50 | –– | NR | –– |
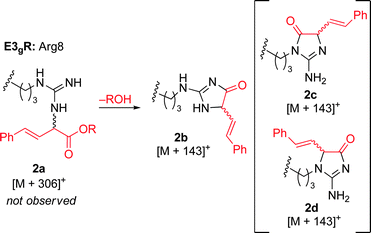
MALDI-TOF MS/MS analysis verifies residue X8 as the site of covalent modification for the major singly modified products (see supporting information†). In the case of E3gW, MS/MS analysis indicates that multiple modifications occur on the same tryptophan side chain. We have been unable to characterize other doubly-modified minor products. The bond connectivity (regio- and chemoselectivity) to the side chain can be inferred from non-aqueous reactions of similar small-molecule substrates,38 but unequivocal determination is challenging: multiple plausible regioisomers are possible in several cases (Chart S1), and epimeric mixtures at the newly-formed, readily enolizeable stereocenter are likely. Table 1 provides a proposed bond connectivity in each amino acid, based on current knowledge. For example, monosubstituted benzene rings (analogous to phenylalanine) have been shown to afford 3,4-cyclopropanation as the major product in dirhodium-catalyzed diazo decomposition reactions.38d In most cases, the product mass indicates a simple adduct of the starting material and the carbene derived from diazo 1. Arginine is the exception: in that case, the product has a mass consistent with loss of the PEG alcohol, likely through formation of an imidazolone after initial N–H insertion (eqn (1)).39 Depending on the regiochemistry of the insertion and cyclization steps, three structural isomers are possible (2b–d). It may be possible to determine the isomeric structure with spectroscopic methods, such as NMR, in ambiguous cases such as arginine modification. However, close analogues of the arginine-derived imidazolones 2b–d have been prepared, and the reported 1H and 13C chemical shifts in the imidazolone ring are nearly identical (Δ∂1H <0.1 ppm and Δ∂13C <5 ppm).39b Because of the expected difficulty in distinguishing among spectroscopically similar species, we have not pursued this line of analysis.
 | (1) |
We have never observed modification of any residue other than tryptophan in non-selective modification,30 making it impossible to quantify the rate enhancement for other reactive residues. In the case of tryptophan, we were able to estimate a proximity-driven rate enhancement of >103. To probe the modification efficiency of the newly reactive residues, a competitive modification reaction was carried out between E3gQ and a tryptophan-containing peptide with randomized sequence, Wrandom.40 After 6 h, full conversion of the glutamine residue in E3gQ was obtained while conversion of the inherently more reactive tryptophan in Wrandom reached only 2% (eqn (2), Fig. S16†). This experiment demonstrates that molecular recognition can be used to override inherent side chain reactivity.
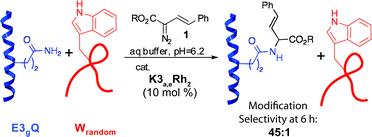 | (2) |
Modification of the natural b-Zip domain, c-Fos
The ability to modify a range of amino acids should enable protein modification without the need to find or engineer a specific residue at the binding site. To explore this concept, we investigated the bZip domain of c-Fos, an oncogene product that regulates transcription through the formation of a heterodimeric coiled-coil with c-Jun.41 Coiled-coils are among the most common biological protein-protein interactions and are analogous to the designed system examined initially. A glutamine residue in a flanking e position (Q19, see Fig. 2A) of the Jun–Fos dimer was identified as a potential target based on structural data.42 We designed a metallopeptide Jun(Rh2), based on an affinity-optimized c-Jun variant,43 with glutamate residues in g and d positions to position the dirhodium center near the target glutamine (Fig. 2A–C).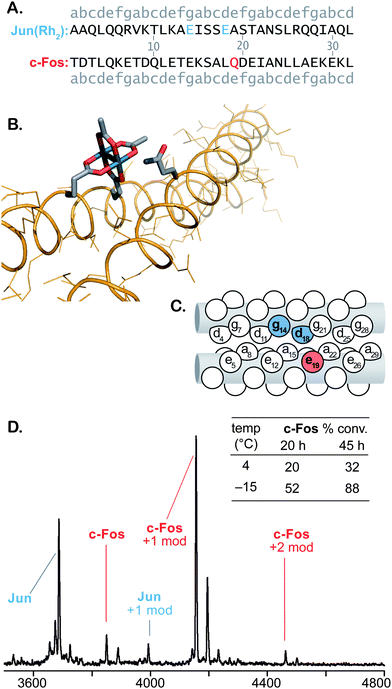 | ||
| Fig. 2 Modification of c-Fos catalyzed by Jun(Rh2). Sequences (A), molecular illustration based on PDB pdb:%3Ca%20href= (B), and “knob-in-hole” diagram (C) illustrate the expected alignment of the catalyst and c-Fos Gln19. MALDI-TOF MS spectrum of a crude reaction and tabulated conversion results (D). All reactions are 100 μM in substrate at pH 6.5. Metallopeptide and diazo 1 were added portionwise to final concentrations of 200 μM and 10 mM, respectively. Reactions at −15 °C contain 20% ethylene glycol cosolvent. See supporting information† for details and MS/MS spectra that indicate Q19 modification. | ||
Employing previously developed procedures,35 the purified Jun variant was metalated with Rh2(OAc)2(tfa)2 in aqueous buffer. The isolation of the Jun(Rh2) metallopeptide proved challenging due to its poor solubility. In initial studies at 4 °C, we found that crude Jun(Rh2) obtained directly from the metalation process could be used in modification reactions—avoiding material loss that accompanied isolation—with activity identical to that of the purified material. Further studies thus utilized unpurified Jun(Rh2).
Initial attempts to carry out covalent modification of c-Fos (100 μM) with 1 equiv Jun(Rh2) at room temperature resulted in 17% conversion after 24 h. Coiled-coils typically become more stable at lower temperatures, so we lowered the temperature of the modification reaction in an attempt to improve catalyst–substrate association. In this case, decreasing the temperature and adding Jun(Rh2) and diazo 1 in small portions afforded 32% conversion in 45 h at 4 °C and 88% conversion over the same time period at −15 °C (Fig. 2D). We also examined the catalytic activity of a second metallopeptide based on the leucine-zipper, Max.44 Although Max is not reported to be a ligand for c-Fos, the metallopeptide derivative Max(Rh2) exhibited catalytic activity comparable to Jun(Rh2), affording 72% and 83% conversion of c-Fos after 45 h at 4 and −15 °C, respectively (Fig. 3D). In both cases, MALDI-TOF conversion analysis was validated by HPLC-MS, which provided comparable results (Figs. S22–23†). Control experiments with Rh2(OAc)4 afforded no observable modification of c-Fos.
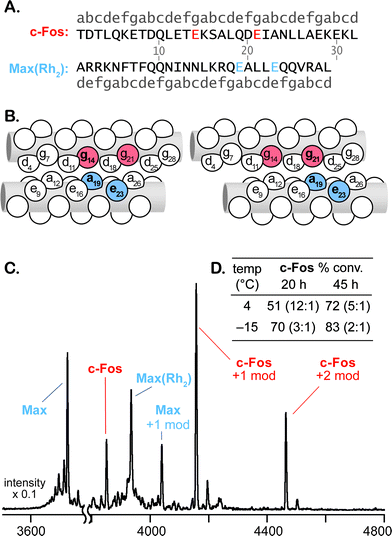 | ||
| Fig. 3 Modification of c-Fos catalyzed by Max(Rh2). Sequences (A) and “knob-in-hole” diagrams (B, C) illustrate the two possible coiled-coil alignments that result in modification of Glu14 (B) and Glu21 (C). MALDI-TOF MS spectrum of a crude reaction and tabulated conversion results (D). Ratios in inset table describe singly:doubly modified product. All reactions are 100 μM in substrate at pH 6.5. Metallopeptide and diazo 1 were added portionwise to final concentrations of 200 μM and 10 mM, respectively. Reactions at −15 °C contain 20% ethylene glycol cosolvent. See supporting information for details†. | ||
MS/MS fragmentation analysis of the reaction products indicates that the two catalysts, Jun(Rh2) and Max(Rh2), modify different c-Fos residues (see supporting information†). For Jun(Rh2), MALDI-TOF MS/MS establishes the expected Gln19 (position e) as the site of c-Fos modification. HPLC-MS indicates the formation of only two singly modified c-Fos products with identical fragmentation patterns (Figs. S22, S25–27†), consistent with the formation of both epimers at the newly-formed stereocenter. The sequence of Max(Rh2) positions the dirhodium core on the opposite face of a coiled-coil with c-Fos. Based on established models for coiled-coil interactions,45 there are two possible orientations of Max(Rh2)–c-Fos, which position the dirhodium core near two different glutamate residues, Glu14 and Glu21 (Fig. 3A–C). For Max(Rh2)-catalyzed modification, the dominant MS/MS fragmentation pathway requires cleavage of the modifying group, establishing a carboxylate side chain as the site of modification (by analogy to similar fragmentation seen in E3gE and E3gD) but preventing an unambiguous determination as to which of the two appropriately positioned glutamate residues is modified (see Fig. 3B–C). The fact that, unlike Jun(Rh2), Max(Rh2) provides significant quantities of doubly modified c-Fos suggests that both Glu14 and Glu21 are modification sites.
The Jun-Fos heterodimer has been studied extensively, and the reported Tm of 16 °C39 provides an ample basis for the interaction of Fos with our Jun variant. (For comparison, a selection of K3a,eRh2–E3gX dimers employed in Table 1 exhibit Tm = 34–47 °C, see Table S-8†). The Jun(Rh2)-catalyzed reaction produces a single glutamine residue (Q19) modification, with sequence specificity that is consistent with that expected from the reported Jun-Fos structure.38 It is difficult to formulate alternative explanations, other than molecular recognition, for the observed results. However, low-affinity interactions like the Jun–Fos dimer are difficult to measure and characterize. The metallopeptide variant, Jun(Rh2), unsurprisingly, has decreased affinity for c-Fos relative to the natural coil. Denaturation experiments monitored by CD spectroscopy (Fig. S-41†) indicate only moderately increased helicity at lower temperatures and provide an upper bound of Tm ≤10 °C for the c-Fos–Jun(Rh2) dimer. As a result of low affinity and factors including low solubility and competing homodimerization, common experimental techniques are not capable of characterizing a c-Fos–Jun(Rh2) dimer in solution. The situation for c-Fos–Max(Rh2) reactions is comparable. Max(Rh2)–c-Fos mixtures display temperature-dependent folding behavior consistent with weak interactions (Tm = 20 °C, Fig. S-42†) that would be necessary for the observed molecular-recognition-based modification.
Conclusions
Our inability to reliably measure the weak affinity of the c-Fos–Jun(Rh2) dimer provides one of the most remarkable and significant insights of this study: templating site-specific reactivity with molecular recognition is possible even on the basis of low-affinity interactions that cannot be characterized by available methods. Successful modification is achieved even though the c-Fos–Jun(Rh2) assembly necessary to template modification is not the major species in solution. This observation expands the generality of our approach: while designing tight ligands for a target protein is often difficult, computational and screening methods routinely deliver modest peptide ligands for a wide variety of protein classes.Although residue-selective chemistries have expanded in recent years,8,9 the range of “targetable” amino acids remains small. Dirhodium metallopeptides represent the only reported method for selective modification of carboxamide (glutamine/asparagine) or phenylalanine side chains. The remarkable amino-acid scope and broad assembly stability requirements enabled by proximity-driven dirhodium catalysis establishes a strategy for shape-selective protein modification, potentially based on any peptide-protein interaction, by combining promiscuous catalytic reactivity with specific molecular recognition. The results presented herein suggest that broader utilization of this catalytic bioconjugation strategy could yield significant opportunities in biochemistry and biomaterials engineering.
Acknowledgements
We thank Jeffrey Hartgerink and Erica Bakota for assistance with peptide synthesis and Christopher Pennington for assistance with mass spectroscopy. We gratefully acknowledge financial support from a J. Evans-Attwell-Welch Post-Doctoral Fellowship (B.V.P.), the Robert A. Welch Foundation research grant C-1680, and Rice University.Notes and references
- C. Walsh, Tetrahedron, 1982, 38, 871–909 CrossRef CAS.
- M. J. Corey and E. Corey, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11428–11434 CrossRef CAS.
- M. Leisola and O. Turunen, Appl. Microbiol. Biotechnol., 2007, 75, 1225–1232 CrossRef CAS.
- L. Baltzer and J. Nilsson, Curr. Opin. Biotechnol., 2001, 12, 355–360 CrossRef CAS.
- H. W. Hellinga, Nat. Struct. Biol., 1998, 5, 525–527 CrossRef CAS.
- D. N. Bolon and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14274–14279 CrossRef CAS.
- A. E. Nixon, M. Ostermeier and S. J. Benkovic, Trends Biotechnol., 1998, 16, 258–264 CrossRef CAS.
- J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253–262 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- C. M. Niemeyer, Bioconjugation protocols: strategies and methods, Humana Press, Totowa, N.J., 2004 Search PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic Press, Boston, MA, 2008 Search PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS.
- S. M. Chen, X. H. Li and H. M. Ma, ChemBioChem, 2009, 10, 1200–1207 CrossRef CAS.
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS.
- C. A. Lewis and S. J. Miller, Angew. Chem., Int. Ed., 2006, 45, 5616–5619 CrossRef CAS.
- K. W. Fiori, A. L. A. Puchlopek and S. J. Miller, Nat. Chem., 2009, 1, 630–634 CrossRef CAS.
- S. J. Miller, Acc. Chem. Res., 2004, 37, 601–610 CrossRef CAS.
- B. R. Sculimbrene and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 10125–10126 CrossRef CAS.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 11653–11656 CrossRef CAS.
- J. M. Xie and P. G. Schultz, Curr. Opin. Chem. Biol., 2005, 9, 548–554 CrossRef CAS.
- R. H. Abeles and A. L. Maycock, Acc. Chem. Res., 1976, 9, 313–319 CrossRef CAS.
- F. Kotzyba-Hibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525–528 CrossRef CAS.
- K. Severin, D. H. Lee, A. J. Kennan and M. R. Ghadiri, Nature, 1997, 389, 706–709 CrossRef CAS.
- A. Saghatelian, Y. Yokobayashi, K. Soltani and M. R. Ghadiri, Nature, 2001, 409, 797–801 CrossRef.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS.
- N. W. Luedtke, R. J. Dexter, D. B. Fried and A. Schepartz, Nat. Chem. Biol., 2007, 3, 779–784 CrossRef CAS.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 438–439 CrossRef CAS.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS.
- B. Apostolovic and H. A. Klok, Biomacromolecules, 2008, 9, 3173–3180 CrossRef CAS.
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8898–8902 CAS.
- A. N. Zaykov, K. R. MacKenzie and Z. T. Ball, Chem.–Eur. J., 2009, 15, 8961–8965 CrossRef CAS.
- A. N. Zaykov, B. V. Popp and Z. T. Ball, Chem.–Eur. J., 2010, 16, 6651–6659 CAS.
- Conversion data are based on MALDI-TOF MS peak intensity. This analytical method was validated for E3gW modification with HPLC using UV detection, which afforded comparable conversion data and deviations ≤10% (ref. 30). For quantitative MALDI-TOF MS analysis of polypeptides, see: J. Sun and B. C. Lynn, J. Am. Soc. Mass Spectrom., 2007, 18, 698–706 Search PubMed; M. P. Stoop, L. J. Dekker, M. K. Titulaer, R. J. Lamers, P. C. Burgers, P. A. Sillevis Smitt, A. J. van Gool, T. M. Luider and R. Q. Hintzen, J. Proteome Res., 2009, 8, 1404–1414 Search PubMed.
- (a) For carboxamide (Gln, Asn), see: R. T. Buck, P. A. Clarke, D. M. Coe, M. J. Drysdale, L. Ferris, D. Haigh, C. J. Moody, N. D. Pearson and E. Swann, Chem.–Eur. J., 2000, 6, 2160–2167 Search PubMed; (b) for sulfhydryl (Cys) see: H. Brunner, K. Wutz and M. P. Doyle, Monatsh. Chem., 1990, 121, 755–764 Search PubMed; (c) for phenol (Tyr) see: D. Haigh, Tetrahedron, 1994, 50, 3177–3194 Search PubMed; (d) for arene (Phe) see: E. Nadeau, Z. J. Li, D. Morton and H. M. L. Davies, Synlett, 2009, 151–154 Search PubMed; (e) for alcohol (Ser), see: G. G. Cox, J. J. Kulagowski, C. J. Moody and E. Sie, Synlett, 1992, 975–976 Search PubMed; D. J. Miller and C. J. Moody, Tetrahedron, 1995, 51, 10811–10843 CrossRef CAS; (f) for carboxylic acid (Asp, Glu), see: S. Bertelsen, M. Nielsen, S. Bachmann and K. A. Joergensen, Synthesis, 2005, 2234–2238 Search PubMed; (g) for indole (Trp), see: J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 Search PubMed.
- We are not aware of reports describing dirhodium-carbenoid reactivity with guanidine substrates. Nevertheless, a small number of cyclic acylguanidines with the substitution pattern described here have been reported. e.g. (a) C. R. Lee and R. J. Pollitt, Tetrahedron, 1970, 26, 3113 CrossRef CAS; (b) N. Ahmed, O. K. Argirov, H. S. Minhas, C. A. A. Cordeiro and P. J. Thornalley, Biochem. J., 2002, 364, 1–14 CAS.
- The Wrandom sequence is: Ac-KQAEIKEEALWQEAAEEKAAQ-NH2.
- E. Shaulian and M. Karin, Nat. Cell Biol., 2002, 4, E131–E136 CrossRef CAS.
- J. N. M. Glover and S. C. Harrison, Nature, 1995, 373, 257–261 CrossRef CAS.
- J. M. Mason, M. A. Schmitz, K. Muller and K. M. Arndt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8989–8994 CrossRef CAS.
- E. M. Jouaux, B. B. Timm, K. M. Arndt and T. E. Exner, J. Pept. Sci., 2009, 15, 5–15 CrossRef CAS.
- A. Lupas, M. Vandyke and J. Stock, Science, 1991, 252, 1162–1164 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data for peptides and metallopeptides, additional MS data for modification reactions, and CD spectroscopic data. See DOI: 10.1039/c0sc00564a/ |
| This journal is © The Royal Society of Chemistry 2011 |