Solid-state dynamic combinatorial chemistry: reversibility and thermodynamic product selection in covalent mechanosynthesis†
Ana M.
Belenguer
*,
Tomislav
Friščić
*,
Graeme M.
Day
and
Jeremy K. M.
Sanders
Department of Chemistry, University of Cambridge, Cambridge, United Kingdom. E-mail: tf253@cam.ac.uk; Fax: +44(0)1223 336 017; Tel: +44(0)1223 763 992amb84@cam.ac.uk.
First published on 5th January 2011
Abstract
We demonstrate the reversibility and thermodynamic control in covalent mechanosynthesis, by using the base-catalysed metathesis of aromatic disulfides as a model reaction. The mechanochemical formation of thermodynamic equilibrium mixtures is observed for both neat and liquid-assisted grinding methodologies. Different methodologies lead to mutually different equilibrium compositions, which also differ from those obtained by solution equilibration. The differences can be explained in terms of crystal packing effects superimposed onto the inherent reactivity of isolated molecules. Calculations indicate that the differences in relative energies of reactants and products in their respective crystal structures can bias the mechanochemical reaction equilibrium towards the complete conversion of reactants into the product, in that way opening the doors for the development of dynamic combinatorial synthesis in the solid state and for the rational design of solid-state synthesis using mechanochemistry.
Introduction
Mechanochemical synthesis is experiencing an intense revival, inspired by the realisation that grinding processes can be significantly accelerated, as well as controlled by small amounts of a liquid phase.1 Liquid-assisted mechanosynthesis has now been applied to the room-temperature construction of porous metal–organic frameworks,2clusters and cocrystals.3 The construction of such materials relies on molecular assemblyviahydrogen bonds,3halogen bonds,4 π⋯π stacking interactions,5 coordination bonds or a combination of these.3,6 These types of molecular self-assembly processes are generally considered to be reversible. Consequently, it is likely that solid-state mechanochemical methodologies could also be successful in the construction of products based on the reversible formation of covalent bonds. Such a possibility is particularly attractive for the room-temperature construction of porous covalent structures, such as capsules or frameworks,7 by supramolecular templating.8 Although covalent mechanosynthesis has been extensively studied,9,10 the research predominantly focused on maximising reaction yields rather than on the underlying thermodynamic factors. Consequently, whereas both kinetically-controlled11 as well as reversible12 reactions were employed, the reversibility of covalent mechanosynthesis has not yet been demonstrated. Similarly, despite its synthetic potential, the possibility of achieving thermodynamic equilibrium in mechanochemical formation of covalent bonds has remained unexplored.We now demonstrate the reversible and thermodynamically controlled formation of covalent bonds under mechanochemical conditions of neat and liquid-assisted grinding (LAG).13 The reversibility of disulfide bond formation, previously investigated in dynamic combinatorial chemistry in solution,14 is now demonstrated under mechanochemical conditions. We also demonstrate that thermodynamic product selection under mechanochemical conditions can lead to a very different outcome than solution synthesis and can be qualitatively explained based on the knowledge of reactant and product crystal structures.
As a model reaction we have explored the reversible disulfide metathesis, which involves the exchange of groups between two homodimeric aromatic disulfides to provide a heterodimer (Fig. 1a,b).15 The metathesis of aromatic disulfides is not spontaneous and usually requires a base catalyst. In dilute solution, the reaction product of all reactions tested is a mixture of the initial homodimers and the resulting heterodimer in the statistical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio. Base-catalysed metathesis of aromatic disulfides in solution was previously described and is complementary to the recently reported phosphine-catalysed metathesis of aliphatic disulfides in solution.16 We selected as reactants the commercially available disulfides containing the 2-nitrophenyl (1), 4-chlorophenyl (2), 4-nitrophenyl (3) and 4-methylphenyl (4) groups (Fig. 1c). The non-symmetrical products are shown in Fig. 1d.
:1:2 ratio. Base-catalysed metathesis of aromatic disulfides in solution was previously described and is complementary to the recently reported phosphine-catalysed metathesis of aliphatic disulfides in solution.16 We selected as reactants the commercially available disulfides containing the 2-nitrophenyl (1), 4-chlorophenyl (2), 4-nitrophenyl (3) and 4-methylphenyl (4) groups (Fig. 1c). The non-symmetrical products are shown in Fig. 1d.
 | ||
| Fig. 1 Schematic representations of: (a) base-catalysed disulfide metathesis (top) with model chromatograms of homodimeric reactants and equilibrium reaction mixture; (b) aromatic disulfide metathesis with dbucatalyst; (c) reactants and (d) heterodimeric metathesis products. | ||
Results and discussion
All mechanochemical reactions were conducted by grinding together a 1:1 stoichiometric mixture of solid reactants, either neat or in the presence of 50 μL acetonitrile (LAG). The total amount of solid reactants was 200 mg in each experiment (for exact weights and molar ratios, see Supplementary Information Table S1†), such that all LAG reactions were conducted at the same liquid-to-solid ratio17 η = 0.25 μL mg−1. As the catalyst, 2 mol % of 1,8-diazabicyclo[5.4.0]undec-7-ene (dbu) was used. Grinding was conducted in a MM200 Retsch mill operating at 30 Hz, using stainless steel jars and two hardened stainless steel 7 mm diameter ball bearings. Care was taken that the overall temperature of the reaction vessel, measured through thermocouples embedded in the grinding jar walls, never exceeded 30 °C. For this purpose, long reaction times were achieved by conducting the reaction in segments of 45 minutes. For analysis, the reactions were quenched with trifluoroacetic acid and analyzed by high-pressure liquid chromatography (HPLC, Fig. 1a) and powder X-ray diffraction (PXRD).
The formation of a solid containing the metathesis product was observed in all reactions (see Supplementary material†). The reaction of 1–1 and 2–2 is particularly interesting: HPLC indicated that the product is almost pure 1–2 (mole fraction of 1–2 in the product: 98%) for both neat grinding and LAG reactions. The mechanochemical formation of a single product is in dramatic contrast to the statistical mixture obtained by solution equilibration, illustrated in Fig. 1a. To verify that the mechanochemical reaction has reached thermodynamic equilibrium, we exploited the fact that the composition of an equilibrium mixture is invariant to initial conditions. This follows from the reversibility of disulfide bond metathesis, which allows the dynamic interconversion of homodimers and heterodimers in the mixture until a stable equilibrium composition is reached. Such equilibration occurs over a range of mixture compositions that can be simplified into five distinct cases (Fig. 2a): pure homodimers (case 1), preferred homodimer formation (case 2), statistical mixture as in solution (case 3), preferred heterodimer formation (case 4) and the pure heterodimer (case 5).
 | ||
| Fig. 2 (a) Schematic chromatograms illustrating five different equilibrating mixtures of homo- (red, blue) and heterodimers (purple). Chromatograms of: (b) a 1:1 mixture of 1–1 and 2–2; (c) same mixture as in (b) after 20 min LAG; (d) same mixture as in (b) after 40 min LAG; (e) statistical mixture of 1–1, 2–2 and 1–2 obtained by solution equilibration; (f) same mixture as in (e) after solvent evaporation and LAG for 40 min. All reactions involved acetonitrile and dbucatalyst. | ||
Neat grinding or LAG of either the solution-equilibrated mixture after solvent removal (which is a case 3 mixture) or of an equimolar mixture of 1–1 and 2–2 (which is a case 1 mixture), yielded the same case 5 product (Fig. 2b–f). Such behavior is consistent with the equilibrium strongly preferring heterodimer formation. Reaction times up to 5 h were explored, revealing that constant composition is achieved within 40 min (Fig. 2b–d).
To verify that the reaction proceeds in the solid, we have used the approach described by Rothenberg et al.:9 to enable direct observation, the reactant mixture was manually ground with and without dbu. Both mixtures remained solid and did not liquify on ageing, dismissing a eutectic-based mechanism. Differential scanning calorimetry of a 1:1 mixture of 1–1 and 2–2 also did not reveal any events other than melting of the reactants. When dbu was present, no thermal event was observed below 67 °C, another indication that the reaction does not involve a bulk eutectic. Similar observations were made for the reaction of 1–1 and 3–3. (See Supplementary Information, Figures S3 and S5†).
The formation of pure 1–2 by LAG was confirmed by the PXRD pattern of the product, which corresponded to the one simulated for the known crystal structure18 (CCDC code FUQLIM, Fig. 3a,b). The PXRD pattern of the neat grinding product was different, suggesting a yet unknown polymorph. Serendipitously, recrystallisation of the ground product from iso-propanol provided single crystals of the new polymorph suitable for X-ray crystal structure determination.‡
 | ||
| Fig. 3 (a) PXRD patterns of products of neat and LAG grinding of 1–1 and 2–2, compared to simulated patterns for two polymorphs of 1–2; (b) fragment of the previously known structure of 1–2 (CCDC code FUQLIM),18 characterised by arrays of C–H⋯O hydrogen bonds19 (C⋯O distances: 3.15Å and 3.21 Å) as the most notable intermolecular contacts; (c) fragment of the structure of the new polymorph of 1–2, displaying chains formed by short Cl⋯O contacts (Cl⋯O distance: 3.23 Å).20 | ||
The PXRD pattern simulated for the new structure matched that of the neat grinding product (Fig. 3a,c). Mechanosynthesis of 1–3 demonstrates similar behavior although the neat grinding equilibration required significantly longer times: the mole fraction of 1–3 rose to 98% within 40 min by LAG, whereas neat grinding resulted in 87% over a period of 14 h (See Supplementary information, Figures S3 and S5†). As further proof that the product of grinding is in dynamic equilibrium, one equivalent of 2–2 was added to two equivalents of the freshly prepared product 1–3. Extended LAG resulted in a complex mixture consisting of 1–1, 2–2, 3–3 as well as 1–2, 1–3 and 2–3, confirming the dynamic nature of the disulfide bonds (Fig. 4a, b).
 | ||
| Fig. 4 Chromatogram of: (a) 1–3; (b) 1–3 after LAG with one-half equivalent of 2–2; (c) 1–2; (d) 1–2 after LAG with one-half equivalent of 3–3; (e) equimolar mixture of 1–1, 2–2 and 3–3; (f) the same mixture after LAG equilibration; (g) the same mixture after equilibration by neat grinding and (h) the same mixture after solution equilibration. | ||
Similar mixtures were obtained by LAG starting from either 1–2 and 3–3, or from equimolar amounts of 1–1, 2–2 and 3–3 (Fig. 4c–f). The formation of the same product from different starting states demonstrates thermodynamic equilibrium. Similar behavior was observed in neat grinding reactions, but led to a different equilibrium composition (Fig. 4g). Both neat grinding and LAG equilibrium mixtures differed from the statistical one obtained in solution (Fig. 4h), again demonstrating that thermodynamic product selection is modified by the mechanochemical environment.
Different behavior is observed in the mechanochemical reactions of 4–4 with 1–1 or 3–3, which result in mixtures containing only 20% of the heterodimer 1–4 or 3–4. Such transformation from a case 1 to a case 2 mixture is also in stark contrast to solution equilibration which yields the statistical case 3 mixture (see Supplementary Information, Figures S6 and S8†). Consequently, the mechanochemical equilibrium now favours homodimers over the heterodimer. This conclusion is supported by using the purified heterodimer 1–4 or 3–4 as the starting material (a case 5 composition). In both cases the reaction induced partial dissociation of the heterodimer to again provide mixtures containing ca. 40% of each homodimer (Fig. 5a,b). Such case 5 to case 2 transformation (Fig. 2a) corresponds to net heterodimer dissociation and covalent self-sorting which contrast the reactions of 1–1 with 2–2 or 3–3 (Fig. 5c,d).
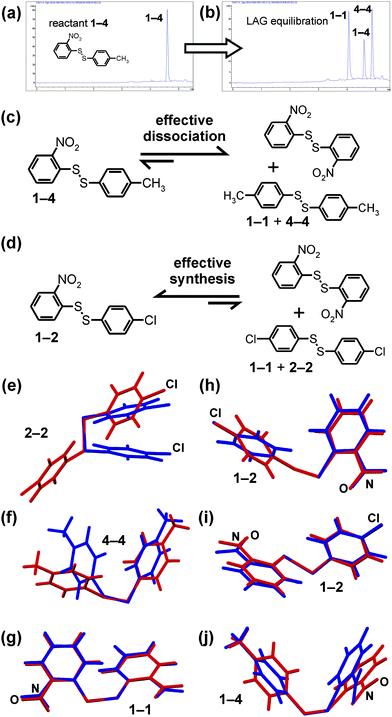 | ||
| Fig. 5 (a) HPLC chromatogram of pure 1–4 (b) HPLC chromatogram of the equilibrium mixture obtained by liquid-assisted grinding of 1–4; (c) the mechanochemical equilibration of solid 1–1, 4–4 and 1–4 favours the dissociation of the heterodimer, in contrast to (d) the system involving solid 1–1, 2–2 and 1–2 which favours heterodimer formation. The comparison of (blue) the calculated lowest-energy gas-phase molecular conformation and (red) the observed conformation in the pure solid for: (e) 2–2 (CCDC code: DCPHDS),22b (f) 4–4;‡ (g) 1–1 (CCDC code: ODNPDS02);18 (h) 1–2 for the new polymorph obtained by neat grinding;‡ (i) 1–2 (CCDC code FUQLIM)18 obtained by LAG and (j) 1–4 (CCDC code: FUQLEI).18 The molecular conformation in solid 2–2 is clearly the most distorted one with respect to the calculated lowest-energy structure. | ||
The equilibration of the reaction mixture suggests that the observed solid-state reactions could be interpreted in terms of relative thermodynamic stabilities of reactant and product crystalline phases. The convergence of different starting states to the same product distribution (Fig. 4) demonstrates the unimportance of kinetic effects related to the crystallisation process itself. Accordingly, a possible explanation for the differences between equilibria in solution and under mechanochemical conditions is that the stabilisation provided by the crystal structures differs significantly between the homodimers and heterodimers. In that way, the energy change for the metathesis reaction involving solid reactants and products could be significantly different from that for molecules in solution. To examine this possibility, theoretical methods were applied to examine the energies of solid reactants and products in reactions of 1–1 with 2–2 and 4–4. Molecular energies were calculated using quantum mechanical (MP2/cc-pVDZ) electronic structure methods. Intermolecular contributions to lattice energies were evaluated using an empirically parameterised atom-atom model potential and atomic multipole electrostatics.21 As a reference, the energies of all isolated molecules (1–1, 2–2, 4–4, 1–2 and 1–4) were calculated in their lowest energy conformation. For 1–1 and 2–2, we find that the total energy of the two non-interacting homodimers is favored over two isolated molecules of 1–2 by 6.7 kJ mol−1. Similarly, the sum of homodimer energies of 1–1 and 4–4 is 5.0 kJ mol−1 lower than for two isolated molecules of the heterodimer 1–4. For calculation of solid-state energies we used the previously reported crystal structures of 1–1 (CCDC code ODNPDS02),18,22a2–2 (CCDC code DCPHDS),22b1–2 (CCDC code FUQLIM)18 and 1–4 (CCDC code FUQLEI)18 as well as the newly determined crystal structure of the neat grinding polymorph of 1–2.
In the 1–1 + 4–4 solid-state system,‡ the total crystal structure energies (evaluated as a sum of the intermolecular interactions in the crystal and the molecular energy of the conformation found in the crystal structure) favor the homodimers by 4.5 kJ mol−1, which is in line with the observed case 2 mixture that results from the mechanochemical reaction. However, for 1–1, 2–2 and 1–2, the balance of energies is shifted towards heterodimers in the solid state: comparison of total crystal energies shows that the polymorph of 1–2 obtained by LAG (FUQLIM) is favored over separate 1–1 and 2–2 crystals by 4.5 kJ mol−1. The calculated energy of the new polymorph of 1–2 obtained by neat grinding is higher than for the LAG polymorph and, in this case, the total energies still favor the homodimers, but only by 1.5 kJ mol−1. For the LAG polymorph, the absolute energy changes agree with the observed preference for heterodimer formation. Although the absolute energy change calculated for the neat grinding polymorph of 1–2 is not in line with the observed equilibrium mixture, the calculated energy change in the solid state is shifted significantly towards the heterodimer, by 5.2 kJ mol−1 compared to the isolated molecules in their most stable conformations.
Overall, the calculated energy changes support the hypothesis that lattice energies are a major factor in shifting the equilibrium relative to reactions in solution. An interesting feature of the theoretical results is that the energetic preferences are not a direct result of stabilisation by intermolecular interactions of homodimers and heterodimers in their crystal structures. In both systems studied, the average intermolecular energy in the homodimer crystals is more stabilizing than in heterodimer crystals. Instead, homodimers are disfavored for the solid state reaction of 1–1 and 2–2 because the homodimer 2–2 is forced into a relatively high energy conformation18,22 in its crystal structure: 12.7 kJ mol−1 above the lowest energy conformation of the isolated molecule (Fig. 5e). In comparison, the solid-state conformations of 1–1 and 1–2 are within 3 kJ mol−1 of the isolated molecule. The different trend in crystal energies in the 1–1 + 4–4 system is largely due to the less strained solid-state conformation of 4–4 (+8.0 kJ mol−1 above the isolated molecule) compared to that of 2–2. These considerations are illustrated in Fig. 5e–j, which clearly demonstrate that the solid-state molecular conformation for 2–2 is the most distorted from the calculated lowest energy conformation compared to any other investigated homo- or heterodimer.
Conclusions
We have demonstrated the reversible behavior and equilibration in mechanochemical reactions. Mechanochemical equilibration led to considerably different outcomes from conventional solution reactions. Whereas the differences between mechanochemical and solution products are typically interpreted in terms of kinetic effects,12 we demonstrate for the first time that covalent mechanosynthesis is also different in terms of thermodynamic control. The crystalline solid state can amplify a particular product and the shift in equilibrium might be predictable by computational approaches if the reactant and product crystal structures are known or could be predicted.23 Such product selection at the levels of covalent and supramolecular chemistry can be modified by liquid-assisted grinding, suggesting a potentially very versatile and computationally predictable approach to conduct dynamic covalent synthesis24 in a solvent-free or highly concentrated environment.Acknowledgements
The Herchel Smith fund is acknowledged for a research fellowship (TF) and EPSRC for financial support (AB, JKMS). GMD is funded by a Royal Society University Research Fellowship. Dr John E. Davies, University of Cambridge, is acknowledged for single crystal X-ray diffraction measurements.Notes and references
- (a) T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC; (b) G. A. Bowmaker, J. V. Hanna, B. W. Skelton and A. H. White, Chem. Commun., 2009, 2168 RSC; (c) K. L. Nguyen, T. Friščić, G. M. Day, L. F. Gladden and W. Jones, Nat. Mater., 2007, 6, 206 CrossRef CAS.
- (a) T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712 CAS; (b) A. Lazuen-Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC; (c) D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 1249 RSC; (d) W. Yuan, T. Friščić, D. Apperley and S. L. James, Angew. Chem., Int. Ed., 2010, 49, 3916 CAS; (e) W. J. Belcher, C. A. Longstaff, M. R. Neckenig and J. W. Steed, Chem. Commun., 2002, 1602 RSC.
- (a) A. N. Sokolov, D.-K. Bučar, J. Baltrusaitis, S. X. Gu and L. R. MacGillivray, Angew. Chem., Int. Ed., 2010, 49, 4273 CrossRef CAS; (b) T. Friščić, R. W. Lancaster, L. Fábián and P. G. Karamertzanis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13216 CrossRef CAS; (c) M. L. Cheney, G. J. McManus, J. A. Perman, Z. Wang and M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 616 CrossRef CAS; (d) J. A. Perman, K. Dubois, F. Nouar, S. Zoccali, L. Wojtas, M. Eddaoudi, R. W. Larsen and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 5021 CrossRef CAS.
- (a) D. Cinčić, T. Friščić and W. Jones, J. Am. Chem. Soc., 2008, 130, 7524 CrossRef CAS; (b) D. Cinčić, T. Friščić and W. Jones, Chem.–Eur. J., 2008, 14, 747 CrossRef.
- (a) G. Koshkakaryan, L. M. Klivansky, D. Cao, M. Snauko, S. J. Teat, J. O. Struppe and Y. Liu, J. Am. Chem. Soc., 2009, 131, 2078 CrossRef CAS; (b) R. Kuroda, T. Sato and Y. Imai, CrystEngComm, 2008, 10, 1881 RSC; (c) P. J. Nichols, C. L. Raston and J. W. Steed, Chem. Commun., 2001, 1062 RSC.
- (a) G. Lapadula, N. Judaš, T. Friščić and W. Jones, Chem.–Eur. J., 2010, 16, 7400 CAS; (b) T. Friščić, E. Meštrović, D. Škalec-Šamec, B. Kaitner and L. Fábián, Chem.–Eur. J., 2009, 15, 12644 CrossRef CAS.
- (a) B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154 CrossRef CAS; (b) M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042 CrossRef CAS; (c) E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672 CrossRef CAS.
- T. Friščić, A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2008, 8, 1605 CrossRef CAS.
- (a) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef CAS; (b) G. Kaupp, M. R. Naimi-Jamal and V. Stepanenko, Chem.–Eur. J., 2003, 9, 4156 CrossRef CAS; (c) A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131 RSC; (d) B. Rodriguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213 CrossRef CAS.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS; (b) J. Mack and M. Shumba, Green Chem., 2007, 9, 328 RSC; (c) J. Mack, D. Fulmer, S. Stofel and N. Santos, Green Chem., 2007, 9, 1041 RSC; (d) D. A. Fulmer, W. C. Sheraouse, S. T. Medonza and J. Mack, Green Chem., 2009, 11, 1821 RSC; (e) A. N. Swinburne and J. W. Steed, CrystEngComm, 2009, 11, 433 RSC; (f) G. Kaupp, CrystEngComm, 2006, 8, 794 RSC.
- (a) T. Szuppa, A. Stolle, B. Ondruschka and W. Hopfe, Green Chem., 2010, 12, 1288 RSC; (b) M. R. Naimi-Jamal, J. Mokhtari, M. G. Dekamin and G. Kaupp, Eur. J. Org. Chem., 2009, 3567 CrossRef CAS.
- (a) R. S. Varma, Green Chem., 1999, 1, 43 RSC; (b) G. Kaupp, M. R. Naimi-Jamal and V. Stepanenko, Chem.–Eur. J., 2003, 9, 4156 CrossRef CAS; (c) D. C. Waddell, I. Thiel, T. D. Clark, S. T. Marcum and J. Mack, Green Chem., 2010, 12, 209 RSC.
- (a) T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621 CrossRef CAS; (b) D. Braga, S. L. Giaffreda, M. Curzi, L. Maini, M. Polito and F. Grepioni, J. Therm. Anal. Calorim., 2007, 90, 115 CrossRef CAS; (c) D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002 CrossRef CAS.
- (a) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590 CrossRef CAS; (b) S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063 CrossRef CAS; (c) W. J. Lee and G. M. Whitesides, J. Org. Chem., 1993, 58, 642 CrossRef CAS; (d) A. L. Kieran, A. D. Bond, A. M. Belenguer and J. K. M. Sanders, Chem. Commun., 2003, 2674 RSC.
- Heterodimeric aromatic disulfides have been investigated in the contexts of supramolecular synthesis and materials chemistry, see: (a) F. M. Tabellion, S. R. Seidel, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2001, 123, 7740 CrossRef CAS; (b) R. Sudharsanam, S. Chandrasekaran and P. K. Das, J. Mater. Chem., 2002, 12, 2904 RSC.
- (a) R. J. Sarma, S. Otto and J. R. Nitschke, Chem.–Eur. J., 2007, 13, 9542 CrossRef CAS; (b) R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603 RSC.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418 RSC.
- C. Glidewell, J. N. Low and J. L. Wardell, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 893 CrossRef.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- V. R. Peddiredi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 987 RSC.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478 RSC.
- (a) J. S. Ricci and I. Bernal, J. Am. Chem. Soc., 1969, 91, 4078 CrossRef CAS; (b) M. R. Spirlet, G. van den Bossche, O. Dideberg and L. Dupont, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 203 CrossRef.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti, J. Jose, S. R. Gadre, G. R. Desiraju, T. S. Thakur, B. P. van Eijck, J. C. Facelli, V. E. Bazterra, M. B. Ferraro, D. W. M. Hofmann, M. A. Neumann, F. J. J. Leusen, J. Kendrick, S. L. Price, A. J. Misquitta, P. G. Karamertzanis, G. W. A. Welch, H. A. Scheraga, Y. A. Arnautova, M. U. Schmidt, J. van de Streek, A. K. Wolf and B. Schweizer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 107 CrossRef.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef; (b) P. T. Corbett, J. Leclaire, L. Vial, J.-L. Wietor, K. R. West, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experiments and computational methods, HPLC chromatograms, PXRD patterns and DSC data for selected materials. Crystallographic data for 4–4 and the new polymorph of 1–2 have been deposited with the Crystal Structure Database. CCDC reference numbers 796996–796997. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00533a |
| ‡ Crystal structure data: Compound 1–2, CCDC code 796996, C12H8ClNO2S2, Mr = 297.8, monoclinic, P21/n, a = 13.4561(3) Å, b = 7.1030(2) Å, c = 13.8346(4) Å, β = 109.73(3)o, V = 1244.7(2) Å3, Z = 4, T = 180 K, R1 = 0.035 for 2211 reflections with I ≥ 2σ(I) and wR2 = 0.096 for all 2786 reflections, S = 1.047; Compound 4–4, CCDC code 796997, C14H14S2, Mr = 246.4, monoclinic, P21, a = 7.6164(2) Å, b = 5.7256(2) Å, c = 14.7508(5) Å, β = 94.881(2)o, V = 640.93(4)Å3, Z = 2, T = 180 K, R1 = 0.038 for 2494 reflections with I ≥ 2σ(I) and wR2 = 0.090 for all 2946 reflections, S = 1.035. |
| This journal is © The Royal Society of Chemistry 2011 |