Synthesis of catalytically active porous organic polymers from metalloporphyrin building blocks†
Abraham M.
Shultz
,
Omar K.
Farha
,
Joseph T.
Hupp
* and
SonBinh T.
Nguyen
*
Department of Chemistry and Institute for Catalysis in Energy Processes, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu; stn@northwestern.edu; Fax: +1 847 491-7713 STN; Fax: +1 847-467-1425 JTH; Tel: +1 847-467-3347 STN Tel: +1 847-491-3504 JTH
First published on 4th January 2011
Abstract
The synthesis of a porous organic polymer (POP) containing free-base porphyrin subunits has been accomplished by the condensation of a bis(phthalic acid)porphyrin with tetra(4-aminophenyl)methane. Metallation by post-synthesis modification affords microporous materials incorporating either Fe or Mn(porphyrins) that have been shown to be active catalysts for both olefin epoxidation and alkane hydroxylation.
Introduction
Microporous materials have attracted considerable recent attention given their potential applications, in catalysis,1 gas storage,2 and chemical separations,3 among many others. In addition to traditional materials such as zeolites, a number of newer classes of microporous materials have been described, including metal-organic frameworks (MOFs),4,5 covalent-organic frameworks (COFs),6–9polymers of intrinsic microporosity (PIMs),10 porous organic polymers (POPs),11,12 and others.13–18 MOFs have been the most widely studied of these newer materials–partly because of their well-defined crystalline structure and partly because of the available enormous variety of candidate organic struts. A great deal of attention has been given to catalytic applications of MOFs,1 due to the potential for integrating the well-defined, single-site activity of homogenous catalysts with the shape-, size-, chemo-, and enantio-selectivity that can be designed into the micropores. However, incorporation of catalytic struts with active metal sites has proven to be a significant challenge due to the difficulty in preventing an erstwhile catalytically active center from acting as a structural node.19 To avoid this complication, the synthesis of all-organic materials, where all the nodes are formed via organic reactions, is an attractive alternative strategy for incorporating catalytically active complexes into porous frameworks.Metalloporphyrins are particularly desirable to use as struts in porous materials given their well-studied catalytic behavior.20–22 Specifically, Fe(porphyrin) and Mn(porphyrin) are analogues of the heme cofactor in the biologically ubiquitous family of cytochrome P450 enzymes, which are responsible for catalyzing a wide variety of oxidation reactions. However, several factors limit the application of these synthetic heme analogues as oxidation catalysts. Most importantly, synthetic metalloporphyrins can rapidly become deactivated, either through the oxidative degradation of the porphyrin ring, or formation of μ-oxo dimers.20,22 Nature avoids these problems by enveloping the heme moiety within a large protein structure, which can also engender selectivity by controlling substrate access to the active site. By incorporating metalloporphyrins within microporous materials, it is hoped that these essential features of biological systems can be mimicked.
Formation of porphyrin-based polymers23 and nanoparticles24,25 have been reported previously, in the context of stabilizing Fe(porphyrin) catalysts in oxidation chemistry; however, examples of porous polymers based on metalloporphyrins are rare. McKeown and coworkers recently reported a condensation synthesis of polymers of intrinsic microporosity (PIMs) that incorporate Co(porphyrin) and Fe(porphyrin) and showed them to be active for the oxidations of cyclohexene and hydroquinone.26–28 These findings motivated us to apply our recently reported modular strategy for synthesizing diimide-linked porous organic polymers (POPs)11,12 to the challenge of constructing diimide-linked POPs containing free-base porphyrin subunits. The resulting highly rigid, all-organic porous materials should be amenable to post-synthesis modification (PSM) to yield a wide range of metalloporphyrin-based materials that are capable of catalyzing chemical reactions.
Results and discussion
Synthesis and characterization of monomers and polymers
The synthesis of POPs previously developed by our group and others takes advantage of the condensation reaction between amines and acid anhydrides to form robust diimide bonds.11,12,15,27Porphyrins can be incorporated into these POP structures by utilizing free-base porphyrins with either pendant amine or acid anhydride groups. To introduce 3-dimensional pores into the POP network, we employed the easily accessible tetraamine monomer 1 and a bifunctional porphyrin monomer with acid anhydride groups. As outlined in Scheme 1, porphyrin 3 can be synthesized in 13% yield in six steps from 4-methylphthalic acid. The nucleophilic deprotection of the phthalate ester in step 5 led to the exchange of p-fluoro groups with hydroxide ions, as previously observed for perfluorophenyl porphyrins,29 to form the bis(phthalic acid)porphyrin 2.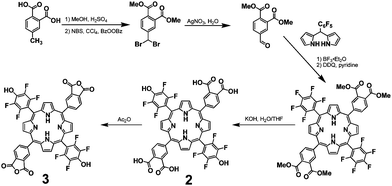 | ||
| Scheme 1 Synthesis of the free-base dianhydride porphyrin monomer 3. | ||
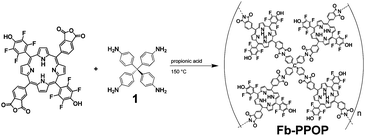
Condensation of the free-base porphyrin monomer (3) with 1 proceeded readily in refluxing propionic acid to give a dark solid that precipitated from solution in 70% yield (eqn (1)). This free-base porphyrin POP (Fb-PPOP) was completely insoluble in common organic solvents. Formation of the diimide linkages in Fb-PPOP was confirmed by FTIR spectroscopy (Fig. 2A) with a new stretch in the carbonyl region at 1724 cm−1. While some residual anhydride bonds are present in the polymeric material, as evidenced by a peak at 1853 cm−1, extending the reaction time did not eliminate them. Thermogravimetric analysis (TGA) of Fb-PPOP after filtration and air-drying confirmed its good porosity, with ∼20% of the initial mass of the sample constituting solvent (Fig. 2C). Evidence for permanent porosity was obtained via gas adsorption measurements (CO2, 273 K) (Fig. 1). Non-local density functional theory (NLDFT) analysis of the CO2 adsorption isotherms of several samples of Fb-PPOP yielded an average surface area of 355 ± 50 m2 g−1.
 | (1) |
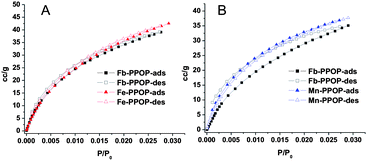 | ||
| Fig. 1 CO2 gas adsorption isotherms of Fb-PPOPs and M-PPOPs, carried out at 0 °C. Each plot include the isotherms for the Fb-PPOP starting materials and the subsequently metallated PPOP. NLDFT surface areas: A) Fb-PPOP: 415 m2 g−1; Fe-PPOP: 453 m2 g−1. B) Fb-PPOP: 376 m2 g−1; Mn-PPOP: 399 m2 g−1. | ||
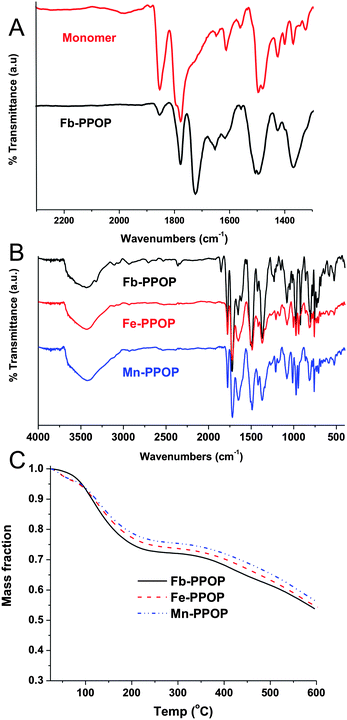 | ||
| Fig. 2 A) FTIR spectra of monomer 3 and Fb-PPOP. B) FTIR spectra of Fb-PPOP, Fe-PPOP, and Mn-PPOP. C) TGA profiles of Fb-PPOP, Fe-PPOP, and Mn-PPOP. | ||
Metallation of PPOPs
Reaction of Fb-PPOP with either anhydrous FeCl2 or MnCl2·4H2O in DMF at 100 °C under nitrogen produced Fe- or Mn-PPOP (eqn (2)). FTIR analysis of the M-PPOPs shows a significant decrease in the intensity of the N–H stretch at 3318 cm−1 upon metallation of the PPOP (Fig. 2B). Analysis with inductively coupled plasma atomic emission spectroscopy (ICP-AES) confirmed metallation of the polymer with up to 3.6 wt% of Fe or 2.8 wt% of Mn. While these are below the theoretical metallation capacities of the Fb-PPOP (4.75 and 4.65 wt% for Fe and Mn, respectively), longer reaction times did not increase the metal content.There are two likely explanations for the aforementioned observed low metal contents in our metallated PPOP materials. Either a significant number of porphyrins remain unmetallated after treatment with the metal salt, or the actual structure of our Fb-PPOP is far from ideal, such that there are fewer available porphyrin sites per mass of the polymer than we expect from an idealized stoichiometry of 2:1 mol mol−13:1. UV-vis analysis would have been a valuable tool for addressing the first hypothesis; unfortunately, the size of our polymer particles (∼100–500 nm) prevented the collection of useful data from diffuse-reflectance measurements. The second hypothesis is more difficult to address given the amorphous, solid-state nature of the PPOPs. We are currently carrying out X-ray scattering experiments in an attempt to interrogate the real structure of these materials; these results will be reported in due course.
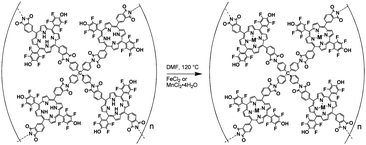 | (2) |
Metallation had little effect on the material's porosity—the CO2 adsorption isotherms for Fe- and Mn-PPOPs are quite similar to that of Fb-PPOP (Fig. 1). TGA indicates that the metallated PPOPs absorb the same amount of solvent as the unmetallated material (Fig. 2C), further supporting this conclusion. The similar porosity of the metallated materials compared to Fb-PPOP additionally suggests that the metal uptake is mostly due to selective metallation of the porphyrin ligands and not adsorption of metal ions within the micropores of the material. In addition, powder X-ray diffraction data of the PPOP materials shows no peaks (see ESI†), suggesting that crystalline metal nanoparticles were not formed during the metallation.
Catalytic activity of Fe- and Mn-PPOP
In the presence of a variety of terminal oxidants, Fe and Mn(porphyrin) complexes are well-known to function as catalysts for a range of oxidation reactions. To test the catalytic activity of the metallated PPOPs (M-PPOPs), we examined the epoxidation of styrene (eqn (3)) by a soluble analogue of iodosylbenzene30 (4, this oxidant is known to be unreactive with olefins in the absence of a catalyst, see ESI† for important safety warnings regarding the synthesis of this oxidant). As a control, these epoxidations were compared against reactions catalyzed by the corresponding metallated meso-tetrapentafluorophenylporphyrin homogenous complexes ((TPFPP)M, M = FeCl, MnCl). At 0.025 mol% catalyst loading, the homogenous catalysts rapidly produce epoxides, but with facile deactivation and degradation of the catalyst (after 780 turnovers in the case of (TPFPP)Mn, and 170 in the case of (TPFPP)Fe (Fig. 3)). In contrast, both the Fe- and Mn-PPOP show greater stability than the analogous homogenous catalysts: Mn-PPOP is active for more than 2000 turnovers without displaying any signs of decomposition. As would be expected for heterogeneous catalysts, M-PPOPs generally display a much slower initial rate than the homogeneous analogs.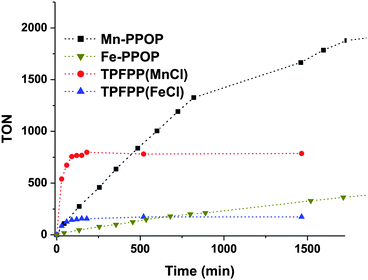 | ||
| Fig. 3 The epoxidation of styrene catalyzed by homogenous M-TPFPP and heterogeneous M-PPOP catalysts (0.025 mol% catalyst, by metal). | ||
While both M-PPOPs can be recycled, their activities are greatly reduced. Fe-PPOP retains only 70% of its initial activity in the second cycle of epoxidation and 23% in the third cycle (Fig. 5). Mn-PPOP also displays a substantial difference between the first and second cycles, with only 60% activity retained in the second cycle. However, after the second cycle, Mn-PPOP shows much less additional degradation than Fe-PPOP, with very similar activity evident for the third and second cycles. The loss of catalytic activity can be attributed to the oxidation of individual pyrrolic rings, not destruction of the polymer framework. While both materials are visibly bleached after a single cycle of catalysis, turning from a dark color into a light tan, SEM analysis of the polymer after bleaching shows identical spherical morphology and particle size (100–500 nm) as the pristine M-PPOPs (Fig. 4). Surprisingly, bleaching is not accompanied by demetallation of the material, as ICP-OES analysis shows similar Fe and Mn contents (See ESI†).
 | (3) |
 | ||
| Fig. 4 SEM images of pristine Fe-PPOP (A) and Mn-PPOP (B) for comparison to after-catalysis Fe-PPOP (C) and Mn-PPOP (D). | ||
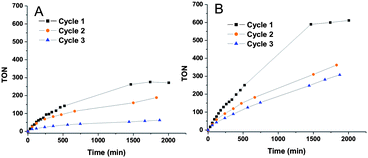 | ||
| Fig. 5 The catalytic recycling profiles of M-PPOP catalysts: A) Fe-PPOP. B) Mn-PPOP. | ||
The new M-PPOPs also proved catalytic for alkane oxidation, albeit with much lower activity than for expoxidation (see Fig. S6 in ESI†), as expected for iodosylbenzene oxidant.22 The oxidation of cyclohexane with 1 mol% of Fe- and Mn-PPOP produced 12% and 48% yields of product (based on oxidant), respectively. In both cases, the product was a mixture of cyclohexanol and cyclohexanone (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in the case of Fe; 95:5 for Mn).
:3 in the case of Fe; 95:5 for Mn).
We note that porphyrin nanoparticles have recently been reported with impressive enhancement of catalytic oxidation activity from the respective building blocks.24,25 However, the manner in which porphyrin molecules are ordered in these nanoparticle materials is also unknown, rendering them difficult to study and improve. While our PPOPs display lower catalytic activity than the aforementioned porphyrin nanoparticles, the modularity of our synthesis strategy lends itself readily for systematic modifications that can aid in future structure-property investigations using solid-state techniques such as density measurements, pore-size distribution analysis, and X-ray scattering. The resulting data can then be used to improve the materials design and catalytic behavior of both types of porphyrin-based materials.
Conclusions
In summary, we have demonstrated a modular method for incorporating metalloporphyrin catalysts as structural components of microporous POPs and demonstrated the feasibility of using these materials as oxidation catalysts that display significantly greater activity than their homogenous counterparts. The ability to prepare porphyrin-based porous organic polymers in free-base form, and then metallate them subsequently, overcomes a problem often encountered with related coordination polymers. Indeed, for the latter, microporous materials featuring coordinatively unsaturated metal sites can be directly synthesized only with appreciable difficulty. Fb-PPOP and similar materials should be modifiable to incorporate nearly any transition metal, as well as many main groups elements, using well-established porphyrin chemistry. Such studies, along with investigations into the catalytic properties of these materials using environmentally friendly reagents and oxidants, will be reported in the near future.Acknowledgements
We thank Mr. Chaiya Prasittichai for performing SEM analysis. SEM measurements were performed in the EPIC facilities of the NUANCE Center at Northwestern University, which is supported by NSF-NSEC, NSF-MRSEC, the Keck Foundation, the State of Illinois, and Northwestern University. NMR, MS, and ICP-OES measurements were performed in the IMSERC facility at Northwestern University, which is supported by NSF (CHE-9871268), the State of Illinois, and Northwestern University. We acknowledge the US Department of Energy (Grant No. DE-FG02-03ER15457), the Northwestern NSEC, AFOSR, and DTRA for funding this study. This work is also supported in part by the Institute for Atom-efficient Chemical Transformations (IACT), an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences.Notes and references
- J.-Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- D. J. Tranchemontagne, J. L. Mendoz-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914 CrossRef CAS.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- O. K. Farha, Y.-S. Bae, B. G. Hauser, A. M. Spokoyny, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Chem. Commun., 2010, 46, 1056 RSC.
- O. K. Farha, A. M. Spokoyny, B. G. Hauser, Y.-S. Bae, S. E. Brown, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Chem. Mater., 2009, 21, 3033 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS.
- P. Kuhn, A. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333 CrossRef CAS.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439 CrossRef CAS.
- R. W. Tilford, W. R. Gemmill, H.-C. z. Loye and J. J. Lavigne, Chem. Mater., 2006, 18, 5296 CrossRef CAS.
- S. Kitagawa, S.-i. Noro and T. Nakamura, Chem. Commun., 2006, 701 RSC.
- B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS.
- Metalloporphyrins in Catalytic Oxidations; R. A. Sheldon, Ed.; Marcel Dekker, Inc.: New York, 1994 Search PubMed.
- M. J. Gunter and P. Turner, Coord. Chem. Rev., 1991, 108, 115 CrossRef CAS.
- T. G. Traylor, Y. S. Byun, P. S. Traylor, P. Battioni and D. Mansuy, J. Am. Chem. Soc., 1991, 113, 7821 CrossRef CAS.
- X. Gong, T. Milic, C. Xu, J. D. Batteas and C. M. Drain, J. Am. Chem. Soc., 2002, 124, 14290 CrossRef CAS.
- G. Smeureanu, A. Aggarwal, C. E. Soll, J. Arijeloye, E. Malave and C. M. Drain, Chem.–Eur. J., 2009, 15, 12133 CrossRef CAS.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782 RSC.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780 RSC.
- P. Battioni, O. Brigaud, H. Desvaux, D. Mansuy and T. G. Traylor, Tetrahedron Lett., 1991, 32, 2893 CrossRef CAS.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, J. Am. Chem. Soc., 1999, 121, 7164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of porphyrin monomer and polymers, a modified synthesis of oxidant 4 and associated safety warnings, as well as a detailed description of catalytic conditions. See DOI: 10.1039/c0sc00339e |
| This journal is © The Royal Society of Chemistry 2011 |