The modified-bead stretched sample method: Development and application to MALDI-MS imaging of protein localization in the spinal cord
Kevin R.
Tucker
,
Leonid A.
Serebryannyy
,
Tyler A.
Zimmerman
,
Stanislav S.
Rubakhin
and
Jonathan V.
Sweedler
*
Department of Chemistry and the Beckman Institute, University of Illinois, 600 South Mathews Ave. 63-5, Urbana, IL 61801, USA. E-mail: jsweedle@illinois.edu; Fax: +217-265-6290; Tel: +217-244-7359
First published on 1st February 2011
Abstract
Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) has been used to create spatial distribution maps from lipids, peptides, and proteins in a variety of biological tissues. MALDI-MSI often involves trade-offs between the extent of analyte extraction and desired spatial resolution, compromises that can adversely affect detectability. For example, increasing the extraction time can lead to unwanted analyte spatial redistribution. With the stretched sample method (SSM), the extraction period can be extended, resulting in reduced analyte redistribution while suppressing detection of cationic salt adducts. The SSM involves thaw-mounting a thin tissue section onto a substrate of small glass beads embedded in Parafilm M and then stretching the membrane to fragment the tissue into thousands of bead-sized pieces. Here, we applied the SSM method to MALDI-MSI using rat spinal cord as a model. We used surface-modified beads coated with trypsin or chymotrypsin in order to facilitate controlled digestion and detection of proteins. The enzymatic reactions were maintained by repeatedly condensing water on the stretched sample surface. As a result, new peptides formed by tryptic or chymotryptic protein digestion were detected and identified using a combination of MALDI-MSI and offline liquid chromatography tandem mass spectrometric analysis. Localization of these peptides indicated the distribution of their proteins of origin, including myelin basic protein, actin beta, and tubulin alpha chain. Additionally, we used uncoated beads to create distribution maps of many endogenous lipids and small peptides. The extension of the SSM using modified beads resulted in the creation of mosaic bead surfaces where adjacent beads were coated with different enzymes or other reactive chemicals, permitting investigation of the distributions of a wider range of analytes in biological samples within a single experiment.
1 Introduction
Mass spectrometry imaging (MSI) of tissue sections, in contrast to other imaging modalities such as immunohistochemistry,1,2 provides chemical and spatial information without requiring analyte preselection. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is a soft ionization technique that produces primarily singly charged molecular ions, allowing for straightforward interpretation of the mass spectra.3 MALDI MS also offers low limits of detection, making it suitable for MSI at high spatial resolutions.4 A number of model neuronal systems have been characterized using MALDI MSI, including the rat spinal cord,5 Jonah crab brain,6Aplysia central nervous system,7 and mouse brain tumors.1 Within these systems, different classes of molecules have been analyzed—lipids,8 peptides,5 proteins,9 and metabolites.10Spatial resolution and analyte extraction can be competing interests in MALDI MSI, with various parameters affecting spatial resolution such as laser spot size and analyte migration and abundance.11 In MALDI MS, a laser is used to ionize analytes directly from a matrix-coated sample surface, so the minimum spatial resolution is defined by the laser spot size, except in cases when the oversampling approach12 or localized application of MALDI matrix are used. More recently, with the incorporation of appropriate optics into the instruments, laser spot sizes are now small enough (≤10 μm) to detect analytes in a single mammalian cell.13 The minimum spatial resolution may be limited by analyte concentration and/or interference during detection from other analytes. An analyte must be present in detectable levels within the effective sample used for analysis; therefore, increasing resolution (decreasing spot size) may bring an analyte below the detection limit. For example, by reducing the illuminated spot size from 200 μm to 10 μm, only 0.25% of the area will be sampled, normally with a concomitant reduction in signal intensity. In most cases, spatial resolution is constrained by the intertwined relationship between sample characteristics/preparation and instrumental figures of merit.
In order to use MALDI for MSI, typically a thin tissue section is collected using a cryostat, thaw-mounted to a conductive surface, and then a matrix is deposited onto the tissue.14 There are a number of ways to apply MALDI matrix onto the sample, either by printing,9 spraying,5 or sublimation.15 Each of these techniques has a number of advantages but represent a compromise between spatial resolution and analyte extraction. The longer a sample is covered with the matrix solution, the larger the fraction of analytes that will be incorporated into the MALDI matrix layer and hence, the more efficiently they will be detected. However, longer extraction times tend to lead to lateral analyte redistribution, thereby reducing the effective spatial resolution obtainable. Several matrix-free approaches alleviate several of these issues with these including laser desorption/ionization16 and nano-assisted laser desorption/ionization,16 although these techniques have been used primarily for analysis of small molecules rather than peptides and proteins. Printing an array of MALDI matrix spots allows for extended analyte extraction times because the matrix spots are separated from each other and so the analytes do not blend together. Due to the drop size of currently available chemical printers,17–19 this approach often results in MSI with a spatial resolution of >100 μm. An alternative is to coat the entire sample surface with a MALDI matrix solution using airbrush- or piezo-assisted spraying, which produces small droplets on impact, forming a thin layer of the solution on the sample surface. Depending on the solvent system used, the solution will evaporate at different rates, with the solvent evaporating more slowly as the fraction of water is increased. Because of the small droplet size, spraying provides a moderate amount of analyte extraction with relatively high spatial resolution and low analyte redistribution. In a recently described, elegant approach known as matrix sublimation, a dry MALDI matrix is applied onto the sample surface,15 and has proven effective for analysis of abundant, low molecular-weight species.20 The stretched sample method (SSM) aims to decouple this relationship between matrix application and the obtainable spatial resolution.21–23
The SSM was originally created as a method to simultaneously prepare many small tissue samples for profiling peptide content.21 Briefly, a thin tissue section is thaw-mounted onto a substrate consisting of ∼38 μm-diameter glass beads that have been embedded in a stretchable membrane. As the membrane is stretched, the tissue section fragments into thousands of bead-sized tissue regions, each spatially isolated by the nonpolar membrane between the beads. The size of the tissue fragmentation is directly linked to the size of the glass beads; the use of larger or smaller glass beads results in larger or smaller tissue fragment pieces, respectively. Furthermore, tissue fragment size is believed to relate only to the size of the bead and to be independent of chemical modifications made to the bead surface, although this was not validated here. Taking advantage of this isolation, samples can be spray coated with matrix and have water condensed on the surface without causing analyte redistribution between the tissue pieces. This allows extended analyte extraction periods, prevents analyte spreading, and even reduces inorganic cation adducts.22,24 Using bright field microscopy and light thresholding to identify the location of each bead, a sample-specific geometry file for automated data acquisition can be generated; a mass spectrum is acquired from each bead location rather than in an ordered pattern. By recording the location where each mass spectrum was acquired, an image can be (re)created from the stretched sample.25 More recently, using several computer algorithms to realign the stretched individual bead/tissue piece positions with their original positions, reconstructed mass spectral images can be generated that map each bead's pre-stretched position in accurate scale and proportion.22
Recognizing the opportunities that the glass bead substrates provide, we chemically modified the bead surfaces to enable a number of applications, including protein investigation. By performing in situ digestion of proteins from stretched tissue samples using enzyme-modified beads, peptides from proteins were identified and localized. Importantly, compared to the larger proteins, peptides are more efficiently ionized and detected. Furthermore, by covalently attaching the enzyme to the beads, we observed a decrease in autolysis, which is assumed to result from steric hindrance due to the enzyme being bound to a solid support.26,27 Additionally, multiple enzyme digestions in situ, as well as analysis of the native tissue, became feasible when using mixed bead substrates consisting of unmodified glass beads as well as enzyme-modified beads. Here, we demonstrate the imaging of lipids, endogenous peptides and proteins via their peptide digestion products from rat spinal cord tissue using the modified bead stretched sample method (MBSSM).
An overview of the MBSSM is depicted in Fig. 1. A substrate is generated by placing 5–10 mg of approximately equal portions of chemically modified and unmodified bead types on top of a square of Parafilm M (25 cm2) and applying heat and pressure to embed the beads into the membrane. After thaw-mounting a thin (14 μm) tissue section, the membrane is stretched over 5-fold in both the x and y directions and then mounted over an indium-tin oxide (ITO)-coated glass slide to reduce sample charging during analysis.22 Individual mass spectra are then collected from each bead position and analyzed according to bead type.
 | ||
| Fig. 1 Schematic showing the modified bead stretched sample method (clockwise from upper left). A substrate is generated by placing 5–10 mg of glass beads, consisting of approximately equal portions of each bead type, on top of a square of Parafilm M (25 cm2) and applying heat and pressure to embed the beads into the Parafilm M (upper left). After thaw-mounting a thin (14 μm) tissue section onto the Parafilm M, the membrane is stretched over 5-fold in both the x and y directions and then mounted over an indium-tin oxide coated glass slide to reduce charging of the sample during MALDI-MSI analysis (right).22 Individual mass spectra are then collected from each bead position and analyzed according to bead type (lower left). | ||
2 Results and discussion
Our goal was to develop an approach that allows a variety of analyte classes to be identified and characterized from a heterogeneous tissue; we selected rat spinal cord as a model because it is biochemically, morphologically, and electrophysiologically well investigated. Its fine structure contains two well-defined regions: the peripheral white matter and central grey matter, two areas that are visually recognizable in optical images of spinal cord sections. The white matter contains primarily myelinated axons and the grey matter contains cell bodies as well as unmyelinated axons.28 Both regions contain characteristic lipids, peptides, and proteins, and play a variety of roles in signal transmission and processing. Although the spinal cord has been analyzed for each of these classes of analytes previously,5,29,30 these three have not been simultaneously investigated within a single experiment. In what follows, we describe the preparation of the enzyme-modified beads, followed by the imaging experiments, and finally, the more traditional tandem-MS (MS/MS) experiments used to identify the compounds detected within the spine.2.1 Confirmation of bead surface-modification
In order to perform in situ digestion of proteins from tissue sections using beads, two proteolytic enzymes, trypsin and chymotrypsin, were covalently attached to the surface of borosilicate glass beads of different colors using the silane crosslinker, 3-(triethoxysilyl)butyl aldehyde (TESBA). The success of the surface modification was confirmed in two ways: solution phase digestion and surface analysis via secondary ion mass spectrometry (SIMS). The solution phase digestion was performed as a qualitative determination of enzyme activity following the bead modification. The enzyme-modified beads were added to separate vials containing a buffered protein solution and incubated over 90 min. Afterwards, MALDI MS analysis (data not shown) of the protein solutions, including a control solution without beads, were performed to ensure that enzymatic digestion was occurring. For a more quantitative analysis of the bead modification, SIMS imaging of the bead surfaces was performed following each step in the synthesis process (Fig. 2). The successful surface modification of the glass beads by TESBA was confirmed on all but one bead (in the sampled region), shown by the decreased Na+ and Cl− signals following the reaction with TESBA (Fig. 2, middle row). Furthermore, enzyme modification of the beads was confirmed by the emergence of CN− signal from the beads following the attachment of the enzyme (in this case, trypsin) to the bead surface (Fig. 2, bottom row). The increase in CN− signal from the enzyme-modified beads indicates that there is more CN−-containing analyte on the bead surface. Since the enzyme was the only CN−-containing species that was applied to the glass beads during modification, this increase in CN− signal indicates successful attachment of the enzyme onto the bead surfaces. | ||
| Fig. 2 Glass beads, prior to modification and following steps (1) and (2), were analyzed using a TOF-SIMS instrument equipped with a 22 keV Au ion source. The images shown here are 500 × 500 μm. The reactions were monitored in positive ion mode by the disappearance of the sodium signal (m/z 23) occurring after the first reaction. There is a single visible bead in the TESBA bead image that was not successfully modified as shown by the remaining sodium signal from its surface. Additionally, a common signal observed from the biological species C2H3O+ (m/z 43), a fragment that arises from the reagents and not the unmodified beads, can be used to monitor the reaction progress. The reactions were also monitored in negative ion mode. The chlorine signal (m/z 35) disappeared after the first reaction, similar to the sodium signal. The CN− (m/z 26) signal is a characteristic signal from proteins having a large number of nitrogen-carbon bonds, and can be used to monitor formation of the protein coating on the bead surface of the trypsin (Trp)-modified beads. | ||
2.2 In situ digestion examined with MSI
In order to compare our approach with a previously published method for performing in situ digestion,9 a Shimadzu chemical inkjet printer (ChIP-1000) was used to apply the matrix. A typical rat spinal cord is approximately 3 mm × 5 mm in cross-section; therefore, spotting at a center-to-center spacing of 250 μm generates an image consisting of 12 pixels × 20 pixels. MS images generated in this fashion are shown in Fig. 3, where various lipids from an undigested sample are shown in panel A and digested protein fragments from a digested sample are shown in panel B. The spatial resolution here is based on the printing of matrix and enzyme. For lipid analysis alone, other matrix application techniques would result in significantly improved spatial resolution. This method of matrix and enzyme application provides several advantages: exact volume control of applied reagents, limited analyte diffusion due to repeated spotting in identical positions, and extended analyte extraction times. This approach for in situ digestion is well suited for mapping larger tissue areas with features that can be resolved at 250 μm spatial resolution. For smaller tissue features, specifically those below 250 μm in size, a higher resolution technique is beneficial. The MBSSM provides this enhancement. | ||
| Fig. 3 MALDI-MS imaging of rat spinal cord. A tissue section was spotted with matrix by a chemical inkjet printer with center-to-center distances of 250 μm, generating an array of 14 pixels × 23 pixels. Masses here are displayed as (M + H)+. (A) Lipids, including phosphatidylcholines (PC) and phosphatidylethanolamine (PE) were mapped via MSI, with the analyte identifications based on previously published data.34(B) Proteins were mapped by performing in situ tryptic digest and detection of resulting peptides as described previously.9 Proteins identified include tubulin alpha chain, glial fibrillary protein (GFAP), actin, neurofilament triplet L protein (NFTLPa), and myelin basic protein. These molecular distribution maps are provided as a reference for comparison of the lipid and protein data presented in this work using the modified bead stretched sample method. | ||
2.3 Modified bead stretched sample method
The MBSSM was applied to rat spinal cord, demonstrating the ability of this method to provide high resolution MS imaging of samples that have relatively small structural features. In order to develop the method and demonstrate its efficacy and reproducibility, this approach was initially used with substrates consisting of a single bead type for each tissue section (data not shown). The data presented here advances beyond a single type of chemical analysis by adding the ability to perform multiple analyses on various small-scale regions of interest within a single tissue section in one experiment. For this proof-of-concept study, unmodified, trypsin-modified and chymotrypsin-modified glass beads of different colors were combined to produce a single substrate. Mass spectra were collected from each individual bead and stored for analysis based on bead type and location. The datasets from each bead type, identified by color, were assembled and analyzed to determine which analytes of interest, identified as described below via liquid chromatography (LC) electrospray ionization (ESI) ion trap (IT) MS/MS, were present in the sample.Following analyte detection and identification (see following section), in-house software was used to generate an ion distribution map in pseudo-color representing the relative ion intensity with respect to the maximum ion intensity observed within that sample for each specified mass-to-charge (m/z) range. Lipid and endogenous peptide ion distribution maps, generated from data collected from the unmodified glass beads, are presented in Fig. 4A. The lipid identifications, based on mass matches to previously published data from the rat nervous system,31–35 are from the analysis of the unmodified beads within the mixed-bead substrate. A variety of distributions are present for lipids, including analytes that are concentrated in the grey and white matter, in addition to several that are present ubiquitously. The identified lipids include those from the phosphatidylcholine and phosphatidylethanolamine families. Additionally, endogenous peptides were identified, using LC-ESI-IT-MS/MS in addition to previously published work,5 and included fragments of myelin basic protein (MBP) and complexin, as well as neuropeptides such as little SAAS and somatostatin. In the spinal cord, MBP is a heterogeneous set of proteins that increases the electrical signal propagation rate along axons; here it is detected via several characteristic ions. Panels B and C in Fig. 4 depict ion distribution maps from digest products that were identified from the trypsin- and chymotrypsin-modified glass beads, respectively. Each analyte was mass-matched to confirm MS/MS data from the LC-ESI-IT-MS/MS experiments discussed below. While each protein identified using the ChIP in situ method was also identified using the MBSSM, the specific ions that were detected varied in several cases. This variation may be due to differences in digestion; for example, steric effects created by attaching trypsin to a solid support.
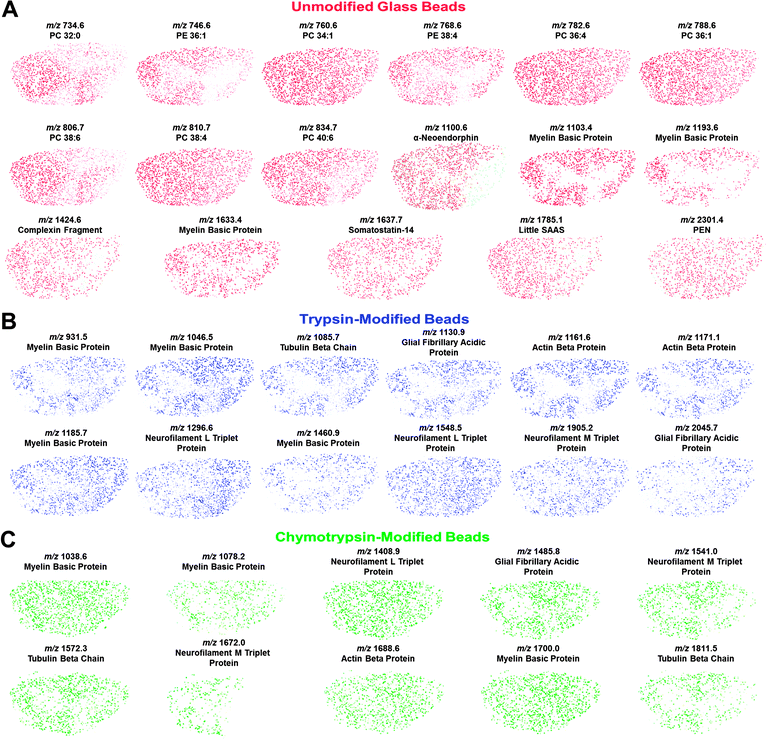 | ||
| Fig. 4 Molecular distribution maps collected from unmodified and trypsin- and chymotrypsin-modified glass beads for peptides detected in spinal cord sections using MALDI-MSI. Masses are (M + H)+. (A) Lipid maps based on mass matches with reported masses34 and maps of endogenous peptides identified using LC-MS/MS and previously reported work.5(B, C) Distribution of peptides formed during protein digest. The peptides are identified by molecular mass matching to the LC-MS/MS data. Overall, the peptide and lipid distributions are consistent among the different bead types and also with previous studies. | ||
After compiling the images from each dataset, ion distribution maps that were related to the same analyte (e.g., MBP) were overlaid, as shown in Fig. 5A. Each color represents a corresponding glass bead analyte signal, red for unmodified, blue for trypsin-modified, and green for chymotrypsin-modified. Fig. 5A details the overlay process for all analyte signals from MBP. By overlaying these maps, an analyte's distribution can be further confirmed because the distributions from each image should correlate. The lipid distributions observed from the unmodified glass beads (individual images shown in Fig. 4A) during the stretched sample analysis were overlaid with signals that were observed in the trypsin- and chymotrypsin-modified glass bead signals (individual images not shown), as shown in Fig. 5B. These overlaid images are intended to provide a total map for each lipid type because the lipids would not be digested on the enzyme-modified beads; thus, for the lipids, the three maps include the same m/z signals. While these images have some variation in distribution by bead type, there is signal from all types of beads throughout each image. Once again, the identified lipids include those from the phosphatidylcholine and phosphatidylethanolamine families. These lipids have been imaged using other mass spectrometric techniques. For example, Monroe et al.5 used SIMS to image the choline head group from phosphatidylcholine and a fragment of phosphatidylethanolamine, both of which were shown to be present in higher levels in the central grey matter than the surrounding white matter. Further, in the reported SIMS results, the observed lipid class distributions of different classes of lipids can be significantly different than the results from examining each individual lipid species. In another study, desorption electrospray ionization (DESI)32 was used to detect many of the same lipid species, but the molecular distributions were not presented. In Fig. 5C, composite images from various protein signals from the spinal cord, including MBP, glial fibrillary acidic protein, and actin beta protein, are displayed. Here, the images consist of multiple overlaid maps from each bead type, and hence correspond to particular analytes. These maps contain different m/z values that are identified as the same analyte (e.g., MBP). Generating composite analyte maps therefore provides an additional degree of certainty in the identification of specific proteins because the overlapping images correlate to each other. An advantage of the MBSSM is its ability to image molecular distributions for proteins, endogenous peptides, and lipids using a single tissue section and a single data acquisition.
 | ||
| Fig. 5 Composite molecular distribution maps collected using the modified bead stretched sample method to analyze rat spinal cord. Composite images are used to provide the summed distribution from all signals detected with three bead types. (A) Schematic showing the process used to generate the composite images. The unmodified, trypsin-modified and chymotrypsin-modified bead images that correlate to a single analyte signal are overlaid, producing a composite image where the red dots represent unmodified glass beads, the blue dots represent trypsin-modified beads, and the green dots represent chymotrypsin-modified beads. (B) Composite images for lipid signals from a trypsin- and chymotrypsin-modified, and unmodified bead substrate that was analyzed using MALDI-MS. Lipid identification was based on mass matches to literature values. (C) Composite images from the same mixed-bead substrate that correspond to the protein signals that were observed. All protein identifications were based on mass matches to LC-MS/MS data. | ||
2.4 Identification of analytes
In order to identify the analytes generated from native spinal cord as well as the in situ digestion of tissue sections by enzyme-modified beads, homogenized spinal cord sections were characterized. Specifically, for digested samples, the extracts were digested in the aqueous phase using trypsin and chymotrypsin. The resulting solutions were concentrated and analyzed using LC-ESI-IT-MS/MS. Partial lists of the identified (via Mascot, http://www.matrixscience.com) trypsin and chymotrypsin digest products are shown in Table 1 and Table 2, respectively. The lists were confirmed by manually examining all Mascot sequence results and comparing them to known compounds previously identified within the spine when possible. It is not surprising that peptides from the most abundant proteins in the spinal cord were detected and identified. These results well match with previously obtained results using different approaches.36–38 Native lipids and peptides were identified using LC-ESI-IT-MS/MS, in addition to comparisons with previously published lipid and peptide identifications.5,31–35| ESI-MS/MS m/z | Theoretical m/z | Sequence | Protein |
|---|---|---|---|
| 931.1 | 930.5 | D.TGILDSIGR.F | Myelin Basic Protein |
| 1046.1 | 1045.5 | DTGILDSIGR | Myelin Basic Protein |
| 1110.0 | 1109.5 | K.GAYDAQGTLSK.I | Myelin Basic Protein |
| 1185.3 | 1184.5 | R.TQDENPVVHF.F | Myelin Basic Protein |
| 1335.1 | 1335.6 | K.YLATASTMDHAR.H | Myelin Basic Protein |
| 1460.2 | 1459.7 | R.TQDENPVVHFFK.N | Myelin Basic Protein |
| 1129.5 | 1129.5 | R.SYASSETMVR.G | Glial Fibrillary Acidic Protein |
| 1245.2 | 1244.6 | R.DNLTQDLGTLR.Q | Glial Fibrillary Acidic Protein |
| 2044.2 | 2044.6 | K.EPTKLADVYQAELRELR.L + pyro - Glu (P) | Glial Fibrillary Acidic Protein |
| 3060.1 | 3059.6 | R.ELQEQLAQQQVHVEMDVAKPDLTAALR.E | Glial Fibrillary Acidic Protein |
| 1161.1 | 1160.6 | K.EITALAPSTMK.I | Actin Beta |
| 1170.0 | 1170.6 | R.HQGVMVGMGQK.D | Actin Beta |
| 1515.6 | 1515.7 | K.QEYDESGPSIVHR.K | Actin Beta |
| 1790.5 | 1789.9 | K.SYELPDGQVITIGNER.F | Actin Beta |
| 1960.0 | 1959.9 | K.YPIEHGIVTNWDDMEK.I + pyro-Glu (P) | Actin Beta |
| 1085.0 | 1084.6 | K.EIIDLVLDR.I | Tubulin Alpha Chain |
| 1701.6 | 1700.9 | R.AVFVDLEPTVIDEVR.T | Tubulin Alpha Chain |
| 1823.2 | 1824.0 | K.VGINYQPPTVVPGGDLAK.V | Tubulin Alpha Chain |
| 1295.2 | 1295.6 | K.NMQNAEEWFK.S | Neurofilament Triplet L Protein |
| 1548.1 | 1547.7 | K.QNADISAMQDTINK.L | Neurofilament Triplet L Protein |
| 1159.3 | 1158.6 | R.LRDDTEAAIR.A | Neurofilament Triplet M Protein |
| 1904.2 | 1903.9 | K.AQVQLDSDHLEEDIHR.L | Neurofilament Triplet M Protein |
| 2588.4 | 2588.3 | R.SNHEEEVADLLAQIQASHITVER.K | Neurofilament Triplet M Protein |
| ESI-MS/MS m/z | Theoretical m/z | Sequence | Protein |
|---|---|---|---|
| 1038.2 | 1037.5 | R.TQDENPVVH.F | Myelin Basic Protein |
| 1078.5 | 1077.6 | D.TGILDSIGRF.F | Myelin Basic Protein |
| 1194.3 | 1193.6 | Q.RTQDENPVVH.F | Myelin Basic Protein |
| 1699.2 | 1698.9 | F.KNIVTPRTPPPSQGK.G + Phospho (ST) | Myelin Basic Protein |
| 1130.5 | 1130.6 | L.ARLEEEGQSL.K | Glial Fibrillary Acidic Protein |
| 1291.4 | 1290.7 | L.RLEAENNLAVY.R | Glial Fibrillary Acidic Protein |
| 1437.1 | 1436.8 | L.RKIHEEEVREL.Q | Glial Fibrillary Acidic Protein |
| 1485.1 | 1484.8 | L.ALDIEIATYRKL.L + Phospho (TY) | Glial Fibrillary Acidic Protein |
| 1822.9 | 1823.9 | L.LEGEENRITIPVQTF.S + Amide (C-term); Phospho (T) | Glial Fibrillary Acidic Protein |
| 1192.5 | 1191.6 | F.IGNSTAIQELF.K | Tubulin Beta Chain |
| 1193.3 | 1192.6 | L.TVPELTQQMF.D | Tubulin Beta Chain |
| 1534.2 | 1533.7 | L.SVHQLVENTDETY.C | Tubulin Beta Chain |
| 1571.3 | 1571.5 | L.THSLGGGTGSGMGTL.L + 3 Phospho (ST) | Tubulin Beta Chain |
| 1811.4 | 1810.6 | F.QLTHSLGGGTGSGMGTL.L + Amide (C-term); Oxidation (M); 3 Phospho (ST); Pyro-glu (N-term Q) | Tubulin Beta Chain |
| 2719.6 | 2720.1 | L.TQQMFDSKNMMAACDPRHGRYL.T + Acetyl (N-term); Amide (C-term); Phospho (STY) | Tubulin Beta Chain |
| 1408.2 | 1407.7 | Y.SSSSGSLMPSLENL.D | Neurofilament Triplet L Protein |
| 1529.3 | 1528.8 | L.RLAAEDATNEKQAL.Q | Neurofilament Triplet L Protein |
| 935.6 | 935.5 | L.REYQDLL.N | Neurofilament Triplet M Protein |
| 1376.5 | 1376.7 | Y.RKLLEGEETRF.S | Neurofilament Triplet M Protein |
| 1540.2 | 1539.9 | M.AIKEEIKVEKPEK.A | Neurofilament Triplet M Protein |
| 1671.2 | 1670.9 | Y.AKLTEAAEQNKEAIR.S | Neurofilament Triplet M Protein |
| 1688.1 | 1687.8 | W.ISKQEYDESGPSIVH.R | Actin Beta Protein |
3 Conclusions
Mammals have anatomically, morphologically, biochemically, and functionally complex nervous systems consisting of millions of neurons making trillions of neuronal synapses.39 The modified bead stretched sample method provides a means to simultaneously investigate a broad range of analytes using MALDI MSI. By digesting proteins on spatially isolated modified beads, multiple analyses were performed in parallel on a single tissue section. Modification of bead surfaces with two or more enzymes that have different cleavage patterns helps to increase the user's confidence in correct analyte identification by detecting and matching the spatial distributions of multiple enzymatically formed analytes on a single substrate. Specifically, here we describe a proof-of-concept experiment demonstrating that the MBSSM can be used to acquire imaging data from lipids, native peptides, and digested proteins in a single experiment. The MBSSM lays the groundwork for future molecular cellular imaging studies by creating a flexible platform that will enable fundamental studies of cell function and cellular interactions, using techniques ranging from mass spectrometry to fluorescence and on-bead immunoassay. We anticipate that this method will be further expanded to incorporate additional imaging modalities.4 Experimental
Chemicals, unless otherwise indicated, were purchased from Sigma-Aldrich, St. Louis, MO and used without further modification.4.1 Bead modification
A method created for the surface modification of glass materials such as slides40 was adapted for use in this work. Borosilicate glass spheres, 38 μm in diameter (Mo-Sci Corporation, Rolla, MO), in separate colors for each bead type (unmodified and two enzyme-modified), were placed in individual vials. The beads were immersed in 15% glacial acetic acid and then shaken at room temperature for 30 min to activate the bead surface. Excess acetic acid was subsequently removed and the beads rinsed with deionized water. Two sets of beads were then subjected to a three-step modification protocol in order to covalently attach the enzymes to their surfaces, as follows: (1) TESBA (United Chemical Technologies, Inc., Bristol, PA), 90%, 0.45 mL per gram of beads, was added to each vial. The reaction proceeded for 5 h on a shaker at room temperature. The TESBA solution was removed and the beads rinsed with deionized water. (2) The enzyme solutions, 5.0 mL of 10.0 mg mL−1 trypsin (Type IX-S from porcine pancreas) or α-chymotrypsin (Type II from bovine pancreas) in 0.10 M phosphate buffer, pH 7.0, were added to the beads. The suspensions were placed on a shaker overnight at 4 °C. (3) The enzyme solution was removed and the beads were passivated with 5.0 mL of 1.0 M glycine solution in 0.10 M phosphate buffer, pH 7.0, and allowed to react on a shaker for 30 min at room temperature. The enzyme-modified beads were stored in a freeze dryer overnight or until dry, and then stored at −80 °C until use.4.2 Secondary ion mass spectrometry of bead surfaces
For these studies, a small scoop each of unmodified beads, modified beads after step (1) and modified beads after step (2) were placed on top of separate pieces of indium foil (Alfa Aesar, Ward Hill, MA) and then sandwiched between two glass slides to embed the beads into the foil. Beads that were not embedded were removed using a stream of nitrogen prior to analysis. A TRIFT III time-of-flight (TOF) secondary ion mass spectrometer (Physical Electronics, Chanhassen, MN) was used to confirm the enzyme attachment to the beads. The mass spectrometer was equipped with a liquid gold metal ion source operating at 22 keV and a primary ion beam was randomly rastered in a 256 pixel × 256 pixel region at 8 kHz and 25-ns pulse width. Total ion doses were below the static limit of 1013 primary ions cm−2. No charge compensation was used. The distance between raster positions determined the resolution of the ion images. Data was analyzed using WinCadence software (Physical Electronics, Chanhassen, MN). The resulting MS images are shown in Fig. 2.4.3 Parafilm substrate preparation
Substrates were created by placing approximately 5–10 mg of the bead type of interest onto a single square of Parafilm M (Pechiny Plastic Packaging, Chicago, IL). For multiple bead types on a single substrate, the beads were placed into an Eppendorf tube in equal amounts and manually shaken for a brief interval to ensure a random bead distribution before placement on the membrane. The Parafilm M with beads was then sandwiched between two glass microscope slides. The beads were spread into a monolayer by moving the top slide in a circular fashion until the bead distribution appeared homogeneous and aggregate. Manual pressure was applied for 30 s to the top slide, and excess beads were removed by a spray of nitrogen gas. To prevent any bead movement during later condensation cycles, the Parafilm M was again placed between the glass slides on a heated aluminium block (∼60 °C) and pressure was applied for 10 s. The substrate was again streamed with nitrogen gas in order to remove any loose beads. Substrates were allowed to cool completely before stretching was attempted. If the substrates were not intended for immediate use, they were stored at −80 °C until use.4.4 Tissue preparation
Long-Evans male and female rats (University of Illinois at Urbana-Champaign) were sacrificed by decapitation and the spinal cord removed according to animal use protocols approved by the University of Illinois Institutional Animal Care and Use Committee, and in accordance with all state and federal regulations. The tissue was microwaved immediately after dissection using a Quasar Lifestyle II microwave, model number MQ 5556AU (Matsushita Electric Industrial Co, Kadoma, Japan) for 15 s at high power. The spinal cord was then flash frozen in liquid nitrogen and stored at −80 °C until use. The spinal cord was mounted to the sectioning stage using deionized water as the embedding media; 14 μm sections were prepared at −20 °C using a Leica CM 3050 S cryostat (Leica Microsystems, Bannockburn, IL). The slices were thaw-mounted onto ITO slides (Delta Technologies, Stillwater, MN) for chemically printed samples and Parafilm M bead substrates for stretched sample analysis. The ITO-mounted samples were stored at −80 °C unless they were to be used immediately, in which case they were dried under nitrogen and then transferred to a ChIP-1000 chemical printer (Shimadzu, Tokyo, Japan). The Parafilm M substrate samples were returned to the cryostat chamber and allowed to reach −20 °C. Substrates was then placed face-down against a clean stainless steel block in the chamber, while pressure was applied using a cold (–20 °C) stainless steel block. A warm stream of air (∼37 °C) was applied to the back of the substrates to thaw the tissue and soften the Parafilm M prior to stretching; this was found to greatly enhance stretching ability. The substrates were then stretched at least 5-fold in each direction for a total increase of greater than 25-fold in overall area. The stretched samples were then placed over ITO slides, which provide the conductive surface necessary for MALDI analysis and the clear transparency needed for bright field microscopy imaging of the samples.224.5 ChIP-1000 tissue preparation
Two rat spinal cord sections mounted on an ITO slide were loaded into the ChIP-1000. An optical image of the spinal cord sections was obtained using the onboard optical scanner at 500 dpi resolution. On one spinal cord section, the ChIP-1000 spotted a solution containing trypsin (0.083 mg mL−1) at 250 μm center-to-center spacing between each spot. 15 nL of trypsin solution was deposited in total on each spot by repeatedly depositing a burst of five 100 pL drops in several iterations across the entire tissue sample; this allowed complete solvent drying to occur following each iteration, thus maintaining droplet isolation. There was a 120 s delay time between printing iterations, which allowed for digestion as well as the evaporation of solvent, preventing droplet pooling. On both rat spinal cord sections, the ChIP-1000 was also used to deposit 10 nL of the MALDI matrix solution containing 2,5-dihydroxybenzoic acid (DHB) (25 mg mL−1) dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–water, spotted at a 250 μm center-to-center spacing between each spot (these spots were printed directly on top of the trypsin spots for the first section) with no delay time between printing iterations needed because the spots were dry prior to the conclusion of each printing pass.
:1 methanol–water, spotted at a 250 μm center-to-center spacing between each spot (these spots were printed directly on top of the trypsin spots for the first section) with no delay time between printing iterations needed because the spots were dry prior to the conclusion of each printing pass.
4.6 MBSSM sample preparation
After the Parafilm M tissue substrates were stretched over the ITO microscope slides, they were placed in a lab-built temperature- and humidity-regulated chamber that has been described previously.22 This system allows for thermal cycling of the sample inside a Plexiglas box. The sample was placed on a Peltier device (Ferrotec, Santa Clara, CA) inside a box attached to a water bubbler filled with 0.050 M phosphate buffer, pH 8.0, and a direct nitrogen line to increase humidity to 85–90%. The samples were monitored in real time using a CCD camera (DFW-X700, Sony, Tokyo, Japan) attached to a 7 × zoom microscope (Edmund Optics, Barrington, NJ). A CN77000 temperature controller (Omega, Stamford, CT) controlled by FTC100 software (Ferrotec, Santa Clara, CA) cycled the sample temperature according to the following protocol: cool to 10 °C over 30 s and hold for 45 s to create droplets of approximately 50 μm around individual beads; warm to the dew point over 90 s and hold for 480 s to provide a buffered aqueous environment for enzymatic digestion to occur; warm to 30 °C over 60 s and hold for 45 s to evaporate all water droplets before the next cooling cycle begins. This protocol was cycled for 3 h. The ambient humidity of the atmosphere was taken into account and temperatures were adjusted accordingly to ensure a well-controlled water droplet radius so as not to flood the sample and allow analyte migration.4.7 Geometry file creation
Following thermal cycling, an UltrafleXtreme MALDI-TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) was used at full laser power with a sampling frequency of 1 kHz to create a grid of small holes in the Parafilm to enable sample alignment and geometric calibration, as previously described.2,25 Subsequently, bright field images were taken with an AxioVert 200 fluorescence light microscope (Carl Zeiss, Inc., Jena, Germany) to generate a mosaic map that was automatically merged to create a high resolution image for geometry file creation. To allow for automated mass spectra acquisition at each bead position, a custom geometry file was created based on the bead locations, as observed in the bright field image, using the holes in the Parafilm M for alignment. Each bead color was isolated using Adobe Photoshop CS4 (Adobe Systems Inc., San Jose, CA) in order to determine the position of each bead type. Image J, version 1.41 (developed at the US National Institutes of Health and available at http://rsb.info.nih.gov/ij/) was used to find bead positions based on radius and circularity restrictions and light threshold parameters. Then, bead geometry files were created by using the Java JDK 1.6 (http://java.sun.com) Applet, available at http://neuroproteomics.scs.illinois.edu/imaging.html, to scale the data accordingly.4.8 Air brush MALDI matrix sample coating
The samples were spray-coated using an airbrush held at a distance of 15 cm with a 30 mg mL−1 solution of DHB in 50:50 acetone/water, propelled by purified nitrogen. Multiple coats were quickly applied until the sample exhibited homogeneous DHB crystal formation, as monitored using a low-power light microscope.
4.9 MALDI mass spectrometry
Mass spectra were collected from each bead position as determined by the custom geometry file in positive ion mode on the UltrafleXtreme MALDI-TOF-TOF mass spectrometer equipped with a frequency tripled Nd:YAG solid state laser. At each bead position, 250 spectra were acquired at 500 Hz, summed, and saved for analysis.4.10 Data processing
Following data acquisition, the mass spectra .fid files were converted to .mzXML using the batch conversion function in CompassXport version 1.3.10 (Bruker Daltonics), followed by conversion to text files using MATLAB, version 7.2 (The MathWorks, Natick, MA) with wrapper code that can be found at http://neuroproteomics.scs.illinois.edu/imaging.html. The mass spectral information was then converted into pseudo-color images for each previously identified analyte signal based on signal intensity within an m/z range. Signals above a minimum defined threshold were mapped using a color having a contrast directly correlated to the absolute intensity normalized to the maximum peak intensity for the specified m/z range of the analyte of interest. The different bead types were plotted in different colors and then overlaid to create a composite image, using Photoshop. The image construction Java script was adapted and modified from the code found at http://neuroproteomics.scs.illinois.edu/imaging.html.4.11 Liquid chromatography (LC) analysis
Chromatographic separation and MS analysis of digested and undigested spinal cord homogenate samples were performed using a capillary LC system (Micromass, UK) coupled to an HCT Ultra ESI-IT-MS/MS mass spectrometer (Bruker Daltonics) in order to identify the native and digested peptides present in each sample. A 1:50 enzyme:substrate mass ratio was calculated using several milligrams of sectioned rat spinal cord. The enzyme was suspended in a 25 mM ammonium bicarbonate solution, pH 8.0, and quickly shaken. The substrate and enzyme solutions were then agitated for 2 h and allowed to react. Undigested samples were treated similarly without the addition of an enzyme to the buffer solution. The samples were centrifuged (5804R multipurpose centrifuge, Eppendorf) at 10000 rpm for 20 min to pellet insoluble species. Then 5.0 μL of the digest solution was manually injected onto a capillary column (PepMap, C18, 3 μm, 100 Å, 300 μm inner diameter, 15 cm, Dionex, Sunnyvale, CA) using a solvent gradient at 2.5 μL min−1 flow rate. The solvent system was composed of two solutions: solution A (95% H2O, 5% acetonitrile (ACN), 0.1% formic acid (FA) and 0.01% trifluoroacetic acid (TFA)) and solution B (5% H2O, 95% ACN, 0.1% FA, and 0.01% TFA). The 50 min gradient LC separation included 6 steps: 97–80% solvent A in 0–5 min (linear); 80–50% solvent A for 5–30 min (linear); 50–20% solvent A for 30–35 min (linear); 20% solvent A for 35–38 min (isocratic); 20–95% solvent A for 38–40 min; 95–97% solvent A for 40–50 min. The ESI-MS data acquisition of selected analytes was performed in a data-dependent manner using the Esquire software (Bruker Daltonics). Following each MS scan, two peptides were selected to be fragmented based on their intensity. Dynamic exclusion of previously selected peaks was used to limit redundant data acquisition over a short period of time. Tandem mass spectra were used for Mascot searches and then sequences were confirmed manually to verify analyte identities using the BioTools software package (Bruker Daltonics).
Acknowledgements
The project described was supported by the National Institute on Drug Abuse under Award Nos. DA017940 and P30DA018310, and by Award No. 5RO1DE018866 from the National Institute of Dental and Craniofacial Research (NIDCR) and the Office of Director (OD), National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIDA, NIDCR or NIH. The authors gratefully acknowledge the assistance of Timothy Spila and Eric Monroe.References
- E. H. Seeley and R. M. Caprioli, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18126–18131 CrossRef CAS.
- T. A. Zimmerman, S. S. Rubakhin and J. V. Sweedler, Methods Mol. Biol., 656, 465–479 Search PubMed.
- M. Karas, M. Glückmann and J. Schäfer, J. Mass Spectrom., 2000, 35, 1–12 CrossRef CAS.
- T. C. Rohner, D. Staab and M. Stoeckli, Mech. Ageing Dev., 2005, 126, 177–185 CrossRef CAS.
- E. B. Monroe, S. P. Annangudi, N. G. Hatcher, H. B. Gutstein, S. S. Rubakhin and J. V. Sweedler, Proteomics, 2008, 8, 3746–3754 CrossRef CAS.
- R. Chen, L. Hui, R. M. Sturm and L. Li, J. Am. Soc. Mass Spectrom., 2009, 20, 1068–1077 CrossRef CAS.
- R. Kruse and J. V. Sweedler, J. Am. Soc. Mass Spectrom., 2003, 14, 752–759 CrossRef CAS.
- R. C. Murphy, J. A. Hankin and R. M. Barkley, J. Lipid Res., 2008, 50, S317–322.
- M. R. Groseclose, A. Malin, M. H. William and M. C. Richard, J. Mass Spectrom., 2007, 42, 254–262 CrossRef.
- S. Khatib-Shahidi, M. Andersson, J. L. Herman, T. A. Gillespie and R. M. Caprioli, Anal. Chem., 2006, 78, 6448–6456 CrossRef CAS.
- D. S. Cornett, M. L. Reyzer, P. Chaurand and R. M. Caprioli, Nat. Methods, 2007, 4, 828–833 CrossRef CAS.
- J. C. Jurchen, S. S. Rubakhin and J. V. Sweedler, J. Am. Soc. Mass Spectrom., 2005, 16, 1654–1659 CrossRef CAS.
- R. B. Cole, Electrospray and MALDI Mass Spectrometry: Fundamentals, Instrumentation, Practicalities, and Biological Applications, John Wiley and Sons Inc., Hoboken, 2010 Search PubMed.
- T. A. Zimmerman, E. B. Monroe, K. R. Tucker, S. S. Rubakhin and J. V. Sweedler, Methods Cell Biol., 89, 361–390 Search PubMed.
- J. A. Hankin, R. M. Barkley and R. C. Murphy, J. Am. Soc. Mass Spectrom., 2007, 18, 1646–1652 CrossRef CAS.
- V. Vidová, P. Novák, M. Strohalm, J. Pól, V.r. Havlíček and M. Volný, Anal. Chem., 2010, 82, 4994–4997 CrossRef CAS.
- J. Stauber, R. Lemaire, J. Franck, D. Bonnel, D. Croix, R. Day, M. Wisztorski, I. Fournier and M. Salzet, J. Proteome Res., 2008, 7, 969–978 CrossRef CAS.
- P. Chaurand, J. L. Norris, D. S. Cornett, J. A. Mobley and R. M. Caprioli, J. Proteome Res., 2006, 5, 2889–2900 CrossRef CAS.
- P. Chaurand, D. S. Cornett and R. M. Caprioli, Curr. Opin. Biotechnol., 2006, 17, 431–436 CrossRef CAS.
- D. C. Schriemer and L. Li, Anal. Chem., 1996, 68, 2721–2725 CrossRef CAS.
- E. B. Monroe, J. C. Jurchen, B. A. Koszczuk, J. L. Losh, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2006, 78, 6826–6832 CrossRef CAS.
- T. A. Zimmerman, S. S. Rubakhin, E. V. Romanova, K. R. Tucker and J. V. Sweedler, Anal. Chem., 2009, 81, 9402–9409 CrossRef CAS.
- J. Wang, R. Chen, M. Ma and L. Li, Anal. Chem., 2008, 80, 491–500 CrossRef CAS.
- E. B. Monroe, B. A. Koszczuk, J. L. Losh, J. C. Jurchen and J. V. Sweedler, Int. J. Mass Spectrom., 2007, 260, 237–242 Search PubMed.
- T. A. Zimmerman, E. B. Monroe and J. V. Sweedler, Proteomics, 2008, 8, 3809–3815 CrossRef CAS.
- K. Kang, C. Kan, A. Yeung and D. Liu, Macromol. Biosci., 2005, 5, 344–351 CrossRef CAS.
- M. Chellapandian, Process Biochem., 1998, 33, 169–173 CrossRef CAS.
- M. Lisbeth, R. N. Jens, T. Yong and P. Bente, J. Comp. Neurol., 2003, 462, 144–152 CrossRef.
- R. R. Landgraf, M. C. Prieto Conaway, T. J. Garrett, P. W. Stacpoole and R. A. Yost, Anal. Chem., 2009, 81, 8488–8495 CrossRef CAS.
- H. Cheng, G. Sun, K. Yang, R. W. Gross and X. Han, J. Lipid Res., 51, pp. 1599–1609 Search PubMed.
- S. N. Jackson and A. S. Woods, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2822–2829 CrossRef CAS.
- M. Girod, Y. Shi, J.-X. Cheng and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2010, 21, 1177–1189 CrossRef CAS.
- J. J. Jones, S. Borgmann, C. L. Wilkins and R. M. O'Brien, Anal. Chem., 2006, 78, 3062–3071 CrossRef CAS.
- S. N. Jackson, H.-Y. J. Wang and A. S. Woods, Anal. Chem., 2005, 77, 4523–4527 CrossRef CAS.
- S. N. Jackson, H.-Y. J. Wang, A. S. Woods, M. Ugarov, T. Egan and J. A. Schultz, J. Am. Soc. Mass Spectrom., 2005, 16, 133–138 CrossRef CAS.
- Q. Ding, Z. Wu, Y. Guo, C. Zhao, Y. Jia, F. Kong, B. Chen, H. Wang, S. Xiong, H. Que, S. Jing and S. Liu, Proteomics, 2006, 6, 505–518 CrossRef CAS.
- T. Ekegren, J. Hanrieder, S.-M. Aquilonius and J. Bergquist, J. Proteome Res., 2006, 5, 2364–2371 CrossRef CAS.
- R. Lemaire, A. Desmons, J. C. Tabet, R. Day, M. Salzet and I. Fournier, J. Proteome Res., 2007, 6, 1295–1305 CrossRef CAS.
- G. M. Shepherd, The Synaptic Organization of the Brain, Oxford University Press, 1998 Search PubMed.
- A. N. Coyne, L. Benner, J. MacMillan, M. Telepchak, United Chemical Technologies, Bristol, Pennsylvania, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |