Selective photocatalytic aerobic bromination with hydrogen bromidevia an electron-transfer state of 9-mesityl-10-methylacridinium ion†
Kei
Ohkubo
a,
Kentaro
Mizushima
a,
Ryosuke
Iwata
a and
Shunichi
Fukuzumi
*ab
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
bDepartment of Bioinspired Chemistry (WCU), Ewha Womans University, Seoul, 120-750, Korea
First published on 17th January 2011
Abstract
Photocatalytic bromination of aromatic hydrocarbons by molecular oxygen with hydrogen bromide occurs efficiently to produce monobrominated products selectively using 9-mesityl-10-methylacridinium ion (Acr+–Mes) as a photocatalyst under visible light irradiation. Both the product yield and selectivity for the bromination of 1,3,5-trimethoxybenzene were 100% with a quantum yield of 4.8%. The photocatalytic turnover number is 900 based on the initial concentration of Acr+–Mes. The reactive radical intermediates involved in the photocatalytic cycle have been successfully detected by laser flash photolysis measurements. The photocatalytic bromination is initiated by photoinduced electron transfer from the mesitylene moiety to the singlet excited state of acridinium ion, which results in formation of the electron-transfer state of Acr+–Mes (Acr˙–Mes˙+), followed by electron transfer from aromatic hydrocarbons to the mesitylene radical cation moiety and electron transfer from the acridinyl radical moiety to O2. The resulting radical cations of aromatic hydrocarbons react with Br− to produce the corresponding monobrominated products selectively.
Introduction
Halogenated organic compounds provide very important synthetic intermediates in organic synthesis for many applications in the fields of material science, chemical industry and medicine.1Bromination of arenes has been one of the most important and fundamental reactions in organic synthesis, providing key precursors for various transformations such as Grignard reactions and Suzuki–Miyaura coupling.1 A number of protocols are available to achieve bromination with elemental bromine and N-bromosuccinimide (NBS), however, they are toxic, hazardous and less selective.2 Extensive efforts have been made to develop an environmentally friendly bromo source that can be used under mild conditions.2–17 Molecular bromine is most frequently used for bromination of organic compounds. To avoid the use of molecular bromine, NBS has been used for bromination, however, molecular bromine is used to prepare NBS. Alternatively, the brominating reagent can be generated by the oxidation of bromide ion where hydrogen peroxide has frequently been used as a green oxidant that produces only water as the side product.2–6 The better oxidant is obviously oxygen. On the other hand, radical cations are known to react with a nucleophile to form various adducts.5,6,16,17 Electrophilic addition of Br− to radical cations would enable selective bromination following electron transfer. However, there has been no report on selective bromination of organic compounds via the electron-transfer oxidation using oxygen as an oxidant.18,19We report herein that 9-mesityl-10-methylacridinium perchlorate (Acr+–Mes)20 acts as an efficient photocatalyst for selective bromination of aromatic hydrocarbons and thiophenes with aqueous HBr as a Br source and O2 as an oxidant under visible light irradiation. The electron-transfer state, Acr˙–Mes˙+, produced upon photoexcitation of Acr+–Mes, has both strong oxidizing and reducing ability, and this enables not only oxidation of alkyl aromatic compounds and thiophenes but also reduction of O2.20–22 The photocatalytic mechanism is clarified by detecting radical intermediates involved in the photocatalytic reactions with use of laser flash photolysis. The present study provides a unique bromination pathway of aromatic hydrocarbons and thiophenes with HBr as a Br source and oxygen as a green oxidant without using a toxic bromine source. It should be noted that bromination of thiophenes is particularly important in the preparation of oligothiophenes and polythiophenes,23–25 which have many applications as conductive, semiconductive, nonlinear optical and liquid crystalline materials.26
Results and discussion
Photocatalytic bromination of trimethoxybenzene with O2
Visible light irradiation of (Acr+–Mes)ClO4− (λmax = 430 nm, 0.20 mM) in O2-saturated acetonitrile (MeCN) containing 1,2,4-trimethoxybenzene (TMB, 4.0 mM) and 50% aqueous HBr ([HBr] = 20 mM, [H2O] = 100 mM) with a xenon lamp attached with a cut-off filter (λ < 320 nm) for 20 min resulted in formation of a brominated product, 2,4,5-trimethoxy-bromobenzene. The bromination was monitored by 1H NMR as shown in Fig. 1. The product was identified and quantified by comparison of the spectra with that of authentic sample. The time profiles conversion of TMB and formation of the corresponding monobrominated product are the same in the photocatalytic reaction (see the supplementary information (ESI) S1†). The overall stoichiometry of the photocatalytic reaction is given by eqn (1).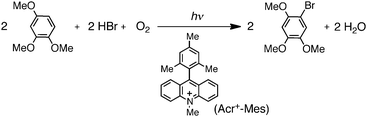 | (1) |
![1H NMR spectra of an oxygen-saturated CD3CN solution containing [Acr+–Mes]ClO4− (2.0 × 10−4 M), 1,2,4-trimethoxybenzene (TMB) (4.0 × 10−3 M) and HBr (2.0 × 10−2 M) (a) before and after photoirradiation [(b) 30 min and (c) 80 min, λ > 320 nm]. Asterisks denote the peaks due to (Acr+–Mes)ClO4−.](/image/article/2011/SC/c0sc00535e/c0sc00535e-f1.gif) | ||
| Fig. 1 1H NMR spectra of an oxygen-saturated CD3CN solution containing [Acr+–Mes]ClO4− (2.0 × 10−4 M), 1,2,4-trimethoxybenzene (TMB) (4.0 × 10−3 M) and HBr (2.0 × 10−2 M) (a) before and after photoirradiation [(b) 30 min and (c) 80 min, λ > 320 nm]. Asterisks denote the peaks due to (Acr+–Mes)ClO4−. | ||
Both the yield and selectivity of the monobrominated product, 2,4,5-trimethoxybromobenzene are 100%, because no further brominated product, dibromo- or tribromo-derivative, was produced as shown in 1H NMR spectra (Fig. 1c). The products and quantum yields were determined by 1H NMR and GC-MS spectroscopies (see the experimental section), which are also listed in Table 1. We have also confirmed that no decomposition of Acr+–Mes occured after the photocatalytic reaction as there was no disappearance of the 1H NMR signal due to Acr+–Mes in Fig. 1.
| Entry | Substrate | Product | Conversion, a% | Yield, a% | Time, h | Quantum yield, b% | E ox , cV |
|---|---|---|---|---|---|---|---|
| a Conditions for photochemical reactions: [substrate] = 4.0 mM; [Acr+–Mes ClO4−] = 0.20 mM; [HBr] = 20 mM; [H2O] = 100 mM. b The quantum yields were determined from the yield of monobrominated product. Experimental error within 0.03%. c Values (versusSCE) are determined by CV and second harmonic ac voltammetry (SHACV). d Minor product is the dibromo compounds. | |||||||
| 1 |

|

|
>99 | >99 | 0.3 | 4.3 | 0.96 |
| 2 |

|

|
>99 | >99 | 0.3 | 4.8 | 1.43 |
| 3 |

|

|
>99 | >99 | 0.3 | 3.9 | 1.49 |
| 4 |

|

|
>99 | >99 | 0.5 | 3.1 | 1.34 |
| 5 |

|

|
>99 | >99 | 1.5 | 1.1 | 1.42 |
| 6 |

|
|
>99 | >99 | 2 | 0.26 | 1.47 |
| 7 |

|

|
>99 | >99 | 16 | 0.01 | 1.76 |
| 8 |
|
|
>99 | 78d | 3 | 0.10 | 1.45 |
| 9 |

|
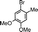
|
>99 | 53 | 2 | 0.36 | 1.33 |
| 10 |

|

|
>99 | 44 | 2 | 0.32 | 1.47 |
Photocatalytic bromination reactions of various aromatic compounds were also examined under the same conditions (Table 1). The efficient catalytic bromination reactions were observed in the case of the methoxy-substituted benzenes. A preparative scale photocatalytic reaction of TMB (200 mg, 1.2 mmol) with 50% aqueous HBr (160 μL 1.5 mmol) in the presence of Acr+–Mes (0.044 mmol) in oxygen-saturated MeCN (100 mL) under photoirradiation by a xenon lamp for 24 h afforded the brominated product in 100% selectivity and in 81% yield (NMR criterion; 61% isolated yield). The isolation procedure is described in the Experimental Section.
When methoxy-substituted aromatic compounds were replaced by toluene derivatives (entries 9, 10 in Table 1), the consumption of substrate occurred efficiently under the same experimental conditions. However, the yield of bromo product and selectivity were significantly lower as compared with methoxy-substituted benzenes, photobromination competes with photooxygenation with oxygen to yield the corresponding aromatic aldehyde.
Photocatalytic bromination of thiophene
Photocatalytic bromination reactions of 3-methylthiophene, bithiophene and trithiophene also occur efficiently with Acr+–Mes and HBr. Visible light irradiation of the absorption band of Acr+–Mes (0.20 mM) in an O2-saturated MeCN solution containing dithiophene (4.0 mM) and HBr (20 mM) for 2 h by xenon lamp results in the selective formation of a monobrominated product [eqn (2)]. No further brominated product, 2,2′-dibromobithiophene was observed under the present experimental conditions. The selective monobromination of 3-methylthiophene also occurred to yield 2-bromo-3-methylthiophene. In contrast to the case of trithiophene, the monobromo product is initially formed together with the small amount of dibromo product. When the starting material was consumed after the photoirradiation for 1.5 h, the yields of mono- and dibromo products are 50% and 50%, respectively. The summary of photobromination of thiophene derivatives is listed in Table 2. | (2) |
| Entry | Thiophene | Product | Conversion, a% | Yield, a% | Time, h | Quantum yield, b% | E ox , cV |
|---|---|---|---|---|---|---|---|
| a Conditions for photochemical reactions: [substrate] = 4.0 mM; [Acr+–Mes ClO4−] = 0.20 mM; [HBr] = 20 mM; [H2O] = 100 mM. b The quantum yields were determined from the yield of monobrominated product. Experimental error within 0.03%. c Values (versusSCE) are determined by CV and second harmonic ac voltammetry (SHACV). d The yield of dibromo product was 50%. | |||||||
| 1 |

|

|
>99 | 81 | 2 | 0.38 | 1.67 |
| 2 |

|
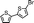
|
>99 | >99 | 1 | 1.3 | 1.23 |
| 3 |

|

|
>99 | 50d | 1.5 | 0.96 | 0.97 |
Mechanism of photocatalytic bromination of substrates with Acr+–Mes, HBr and O2
The photodynamics of the photocatalytic bromination of TMB with Acr+–Mes, HBr and O2 was examined by nanosecond laser flash photolysis. A transient absorption spectrum observed after the laser pulse excitation at 430 nm of an MeCN solution of Acr+–Mes is assigned to the electron-transfer state of Acr+–Mes (Acr˙–Mes˙+: λmax = 500 nm) produced by intramolecular electron transfer from the mesitylene (Mes) moiety to the singlet excited state of acridinium (Acr+) moiety (Fig. 2, blue).20 The broad absorption band in the near IR region (800–1100 nm) is attributed to the π-dimer of Acr˙–Mes˙+ with the ground state of Acr+–Mes.27,28 In the presence of TMB, a new band at 450 nm appeared at 12 μs, which is assigned to the TMB radical cation (TMB˙+)29 in Fig. 2. The rise time profile for formation of TMB˙+ is monitored at 450 nm as shown in Fig. 3a, which obeys pseudo-first-order kinetics. The rate constant for formation of TMB˙+ (krise) increases linearly with increasing concentration of TMB. The second-order rate constant of electron transfer from TMB to the Mes˙+ of the electron-transfer (ET) state of Acr+–Mes was determined from the slope of the linear plot of kriseversus [TMB] to be kTMB = 2.0 × 1010 M−1s−1 (Fig. 3b). The absorption band of TMB˙+ disappeared with second-order kinetics via back electron transfer from the Acr˙ moiety of the ET state to TMB˙+ (Fig. 4a). The decay of TMB˙+ was accelerated by addition of HBr. The decay time profile obeys first-order kinetics in the presence of HBr. The observed decay rate constant (kdecay) increases linearly with increasing concentrations of HBr as shown in Fig. 4b. Thus, TMB˙+ efficiently reacts with Br− to form the Br adduct radical [TMB(Br)˙]. The rate constant for the addition of Br− was determined from the slope of kdecayversus [HBr] to be kHBr = 1.4 × 108 M−1s−1 (Fig. 4b). The electron-transfer oxidation of Br− in the photocatalytic reaction was also examined by quenching of Acr˙–Mes˙+ by HBr. The quenching rate constant of Acr˙–Mes˙+ with HBr is two orders of magnitude smaller (2.9 × 108 M−1s−1) than the kTMB value (Fig. 3b). Thus, the substrate (4.0 mM) rather than Br− (20 mM) is preferentially oxidized under the present experimental conditions.30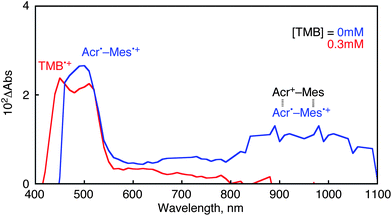 | ||
| Fig. 2 Transient absorption spectra of the ET state of Acr+–Mes (0.2 mM) in the absence and presence of TMB (0.3 mM) in deaerated MeCN after nanosecond laser excitation at 430 nm. | ||
![(a) Transient absorbance time profiles at 450 nm due to TMB˙+ for electron transfer from TMB (0–0.30 mM) to Acr˙–Mes˙+ in MeCN after nanosecond laser flash photolysis (λex = 430 nm); (b) plot of kriseversus [TMB] or [HBr] for the reaction of Acr˙–Mes˙+ with TMB or HBr.](/image/article/2011/SC/c0sc00535e/c0sc00535e-f3.gif) | ||
| Fig. 3 (a) Transient absorbance time profiles at 450 nm due to TMB˙+ for electron transfer from TMB (0–0.30 mM) to Acr˙–Mes˙+ in MeCN after nanosecond laser flash photolysis (λex = 430 nm); (b) plot of kriseversus [TMB] or [HBr] for the reaction of Acr˙–Mes˙+ with TMB or HBr. | ||
![(a) Transient absorbance time profiles at 450 nm due to TMB˙+ in the absence and presence of HBr (0–0.50 mM) in O2-saturated MeCN containing Acr+–Mes (0.2 mM) after nanosecond laser flash photolysis (λex = 430 nm); (b) plot of kdecayversus [HBr] for the reaction of TMB˙+ with HBr.](/image/article/2011/SC/c0sc00535e/c0sc00535e-f4.gif) | ||
| Fig. 4 (a) Transient absorbance time profiles at 450 nm due to TMB˙+ in the absence and presence of HBr (0–0.50 mM) in O2-saturated MeCN containing Acr+–Mes (0.2 mM) after nanosecond laser flash photolysis (λex = 430 nm); (b) plot of kdecayversus [HBr] for the reaction of TMB˙+ with HBr. | ||
The one-electron oxidation potentials (Eox) of aromatic compounds in deaerated MeCN are listed in Table 1. The Eox values (0.96–1.76 V) are lower than the one-electron reduction potential of the ET state of Acr+–Mes (Acr˙–Mes˙+; Ered = 2.06 V versusSCE in MeCN).21 Thus, electron transfer from an aromatic compound such as TMB to Acr˙–Mes˙+ is energetically favorable, whereas electron transfer from toluene (Eox = 2.20 V)31 and benzene (2.32 V)31 to the Mes˙+ moiety is energetically unfavorable. Thus, no photocatalytic bromination of benzene or toluene occurs under the present experimental conditions. The decreased reactivity of methoxybenzene (entry 7 in Table 1) is attributed to the high Eox value (1.76 V). On the other hand, the one-electron reduction of O2 by the ET state of Acr+–Mes in the presence of an acid is known to occur efficiently, producing HO2˙.32 The rate constant of electron-transfer reduction of O2 (kO2) has been reported to be 6.8 × 108 M−1s−1.21
The photocatalytic reaction is initiated by intramolecular photoinduced electron transfer from the Mes moiety to the singlet excited state of the Acr+ moiety of Acr+–Mes to generate Acr˙– Mes˙+ as shown in Scheme 1. The Mes˙+ moiety can oxidize TMB to produce TMB˙+, whereas the Acr˙ moiety can reduce O2 to HO2˙. The TMB˙+ produced reacts with Br− to form the Br adduct radical, which undergoes dehydrogenation with HO2˙ to afford the corresponding brominated product and hydrogen peroxide. Hydrogen peroxide further reacts with HBr and the substrate to produce another brominated product and H2O.33,34
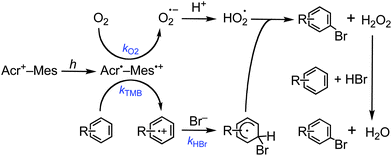 | ||
| Scheme 1 Proposed photocatalytic reaction mechanism. | ||
The quantum yield of photocatalytic bromination of TMB with Acr+–Mes and HBr remains constant (4.3%) with increasing the concentration of TMB and the light intensity under the present experimental conditions (see Fig. 5). This suggests that no radical chain process is involved in the present photocatalytic aerobic bromination of substrates with HBr.
![(a) Dependence of the quantum yields (Φ) on [TMB] for photocatalytic bromination of TMB with Acr+–Mes (0.2 mM) and HBr (4.0 mM) in O2-saturated CD3CN. Light intensity: 0.38 klx. (b) Dependence of the quantum yields (Φ) on excitation light intensity for photobromination of TMB (4.0 mM) with Acr+–Mes (0.2 mM) and HBr (4.0 mM) in oxygen-saturated CD3CN. Excitation wavelength: λ = 430 nm.](/image/article/2011/SC/c0sc00535e/c0sc00535e-f5.gif) | ||
| Fig. 5 (a) Dependence of the quantum yields (Φ) on [TMB] for photocatalytic bromination of TMB with Acr+–Mes (0.2 mM) and HBr (4.0 mM) in O2-saturated CD3CN. Light intensity: 0.38 klx. (b) Dependence of the quantum yields (Φ) on excitation light intensity for photobromination of TMB (4.0 mM) with Acr+–Mes (0.2 mM) and HBr (4.0 mM) in oxygen-saturated CD3CN. Excitation wavelength: λ = 430 nm. | ||
Selectivity of photocatalytic bromination
The selectivity of monobromination is controlled by the difference between the electron-transfer oxidation reactivity of brominated and unbrominated benzenes with the Mes˙+ moiety of the ET state and also by the reactivity of the radical cations with Br−. The one-electron oxidation potential of bromobenzene is lower than that of the benzene.35 Thus, the electron-transfer oxidation reactions of brominated benzenes with the Mes˙+ moiety of the ET state may be as efficient as those of unbrominated benzenes. Thus, the lower reactivity of brominated benzenes as compared with unbrominated benzenes in Table 1 may result from the lower reactivity of the radical cations of brominated benzenes. The atomic charges of the neutral and the radical cation of TMB are shown in Fig. 6, which are estimated by DFT calculations using the B3LYP/6-31+G(d,p) basis set. The position of the largest positive charge on the neutral aromatic compound is the same as that of the radical cation. This indicates that the nucleophilic bromination of the photo-generated radical cation species with Br− results in the same regioselectivity as that for the ordinary electrophilic bromination.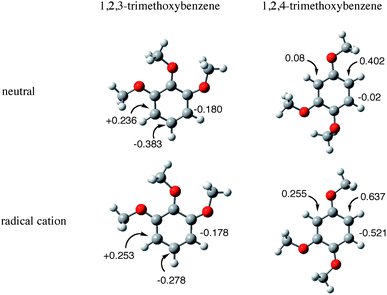 | ||
| Fig. 6 Atomic charges on carbon atoms of 1,2,3- and 1,2,4-trimethoxybenzene of the neutral and radical cation forms calculated at the B3LYP/6-31+G(d,p) basis set. | ||
The determining steps of quantum yield for the bromination are electron-transfer oxidation of the substrate and electron-transfer reduction of O2 with the ET state of Acr+–Mes. The quantum yield of photobromination depends on the electron-transfer oxidation reactivity of substrates with the Mes˙+ moiety and the reactivity of the radical cation with Br−. The aromatic compounds without electron-donating substituent(s) have high oxidation potentials to afford the poor reactivity of electron-transfer oxidation. The higher is the Eox value of substrate, the slower is electron-transfer rate, resulting in the lower quantum yield as shown in Table 1. The significant difference in the reactivity between 1,2,3- and 1,3,5-trimethoxybenzenes despite virtually the same Eox values may result from the energy difference between the radical cation and brominated neutral radical, which are estimated by DFT calculations at the B3LYP/6-31G(d) basis set (Table 3). The Br− adduct of the radical cation of 1,3,5-trimethoxybenzene is 8.5 kcal mol−1 more stable than that of 1,2,3-trimethoxybenzene.
| Substrate | Radical cation | Br adduct radical | Energy differenceb |
|---|---|---|---|
| a Calculated at the B3LYP/6-31G(d) basis set. b Normalized to −2571.9540161 hartree. | |||
| 1,2,3-Trimethoxybenzene | −575.5437207 | −3147.5041129 | 8.5 kcal mol−1 |
| 1,3,5-Trimethoxybenzene | −575.550096 | −3147.504112 | 0 |
When Acr+–Mes was replaced by 9-mesityl-2,7,10-trimethylacridinium ion (Me2Acr+–Mes)21 as a photocatalyst which has a stronger reducing ability at the ET state (Eox = −0.67 V versusSCE) compared to Acr+–Mes, the photocatalytic reaction rate of photobromination of TMB was slower than that with Acr+–Mes. The yield of bromination of TMB was 38% with 37% consumption of substrate after the photoirradiation for 0.3 h (100% for Acr+–Mes). The low yield of bromination catalyzed by Me2Acr+–Mes is attributed to the lower quantum yield of the ET state of Me2Acr+–Mes as compared with Me2Acr+ (vide infra). The quantum yield of the ET state was obtained from the fluorescence lifetime measurements and the comparative method by time-resolved laser flash photolysis.36 The fluorescence decays of Me2Acr+–Mes and Me2AcrH+ are shown in Fig. 7. The lifetimes were determined to be 15 ns for Me2Acr+–Mes and 37 ns for 2,7,10-trimethylacridinium (Me2AcrH+). The fluorescence quantum yields were determined to be 0.21 and 0.50 for Me2Acr+–Mes and Me2AcrH+, respectively. Thus, 60% of fluorescence of Me2AcrH+ was quenched by intramolecular electron transfer from the Mes moiety to the singlet excited state of Me2AcrH+ to form the ET state. The quantum yield of the ET state was directly determined from the transient absorption spectrum of the ET state of Me2Acr+–Mes and the molar absorption coefficient of one-electron reduced Me2Acr+–Mes (Me2Acr˙–Mes, ε = 5000 M−1 cm−1 at 520 nm in MeCN) obtained from electron-transfer reduction of Me2Acr+–Mes with tetramethyl-p-benzosemiquinone radical anion. The quantum yield of the ET state was 60% in MeCN, which is significantly smaller than the value of Acr+–Mes (98%).20a
 | ||
| Fig. 7 Fluorescence decay curves of Me2Acr+–Mes (5.0 × 10−5 M, blue) and Me2AcrH+ (5.0 × 10−5 M, black) in deaerated MeCN. Excitation wavelength: 430 nm. Monitoring wavelength: 510 nm. Inset: First-order plots. | ||
In contrast to the case of Acr+–Mes, the photocatalytic bromination of TMB with O2 in the presence of 9-phenyl-10-methylacridinium ion (Acr+–Ph) proceeds viaelectron transfer from TMB to the singlet excited state of Acr+–Ph (1Acr+–Ph*). The reactivity was particularly decreased because Acr+–Ph does not have a reducing ability at the photoexcited state. The yield of bromination was 10% with 0.6% quantum yield for 0.3 h photoirradiation. The lifetime of 1Acr+–Ph* (τ = 1.5 ns in MeCN)37 is much shorter than that of the ET state of Acr+–Mes. A high concentration of substrate is required to quench the short-lived 1Acr+–Ph*.
We have also examined photocatalytic reactions of mesitylene or durene in with Acr+–Mes and HBr in O2-saturated MeCN. The one-electron oxidation of durene and mesitylene with the Mes˙+ moiety are thermodynamically possible, because the Eox values of the substrates (1.98 V versusSCE for mesitylene; 1.63 V for durene)31 are lower than the Ered value of Mes˙+ (2.06 V).21 However, no bromination occurred under the present photocatalytic conditions. The deprotonation of the radical cation of toluene derivatives such as mesitylene and durene occurs more efficiently to yield the benzyl radical. This is followed by rapid O2 addition to afford the peroxyl radical, leading to the corresponding aldehyde.21 Thus, the main products in photoreaction of mesitylene and durene with HBr in the presence of O2 for 1.5 h were 3,5-dimethylbenzaldehyde (16% yield) and 2,4,5-trimethylbenzaldehyde (10%), respectively.
Summary
The photocatalytic bromination reaction proceeds via intramolecular photoinduced electron transfer from the Mes moiety to the singlet excited state of the Acr+ moiety of Acr+–Mes to generate Acr˙–Mes˙+, followed by electron transfer from the substrates to the Mes˙+ moiety of Acr˙–Mes˙+ and also electron transfer from the Acr˙ moiety to O2 with H+ to produce HO2˙, accompanied by regeneration of Acr+–Mes. The resulting substrate radical cation reacts with Br− to produce the Br adduct radical, which undergoes dehydrogenation with HO2˙ to yield the corresponding brominated product and hydrogen peroxide. H2O2 reacts with HBr and the substrate to yield another brominated product and H2O. Thus, the use of Acr+–Mes as a photocatalyst has enabled us to accomplish the selective photobromination of aromatic hydrocarbons and thiophenes with HBr as a Br source and oxygen as a green oxidant without using a toxic bromine source. Although the substrates that can be brominated are limited by their oxidation potentials, this limitation is compensated for by the high selectivity for the bromination to avoid over-bromination. Such a selective photocatalytic bromination reaction by the present method viaelectron transfer can allow us the large-scale synthesis without the cost of isolation and purification.Experimental
Materials
Chemicals were purchased from a commercial source and used without further purification, unless otherwise noted. 9-Mesityl-10-methylacridinium perchlorate [(Acr+–Mes)ClO4−], 9-mesityl-2,7,10-trimethylacridinium perchlorate [(Me2Acr+–Mes)- ClO4−] and 9-phenyl-10-methylacridinium perchlorate [(Acr+–Ph)ClO4−] were commercially obtained from Tokyo Chemical Industry, Japan. Acetonitrile was of spectral grade, obtained commercially and used without further purification.Reaction procedures and isolation of 2,4,5-trimethoxybromobenzene
The photocatalytic reactions were carried out by the following procedure. Typically, a CD3CN solution (0.6 mL) containing (Acr+–Mes)ClO4− (0.20 mM) and 1,2,4-trimethoxybenzene (TMB, 4.0 mM) in an NMR tube with a rubber septum was saturated with oxygen by bubbling oxygen through a stainless steel needle for 5 min before adding HBr (20 mM). The solution was then irradiated with a 500 W xenon lamp (Ushio Optical ModelX SX-UID 500XAMQ) through a color glass filter that cuts off light of λ < 320 nm at 298 K. After photoirradiation, the corresponding brominated product was identified and quantified by comparison of the 1H NMR spectra with that of an authentic sample and an internal standard. The chemical shifts of all products are listed in ESI S2.†A brominated product, 2,4,5-trimethoxybromobenzene was isolated by the following procedure. Typically, (Acr+–Mes)ClO4− (18 mg, 0.044 mmol) and TMB (0.2 g, 1.2 mmol) were dissolved in an MeCN solution (100 mL) containing 50% aqueous HBr (160 μL, 1.5 mmol). The solution was stirred under photoirradiation through the glass of an aqueous solution of copper sulfate as a color filter (λ = 320–600 nm) at 298 K for 24 h. The reaction mixture was neutralized by adding aqueous NaOH, then diluted with hexane (100 mL) and distilled water (100 mL). The organic layer was washed three times with water (50 mL × 3) and then dried using Na2SO4. The isolated yield of 2,4,5-trimethoxybromobenzene was 61% (0.18 g, 0.73 mmol).
GC-mass measurements
GC-mass measurements were carried out with a Shimadzu GC-17A gas chromatograph and QP5000 mass spectrometer (electron ionization method) which was equipped with a flame ionization detector and a capillary column (122–5532, DB-5MS), 30 m × 0.25 mm. Helium was used as a carrier gas.Quantum yield determinations
A standard actinometer (potassium ferrioxalate)38 was used for the quantum yield determination of the photocatalytic bromination of organic substrates with HBr in the presence of Acr+–Mes in O2-saturated MeCN. Typically, a square quartz cuvette (10 mm i.d.), which contained a deaerated CH3CN solution (3.0 cm3) of Acr+–Mes (2.0 × 10−4 M) and 1,2,4-trimethoxybenzene, was irradiated with monochromatic light of λ = 430 nm from a Shimadzu RF-5300PC fluorescence spectrophotometer. Under the conditions of actinometry experiments, the actinometer and Acr+–Mes absorbed essentially all incident light of λ = 430 nm. The light intensity of monochromatized light of λ = 430 nm was determined to be 2.7 × 10−8 einstein s−1. The quantum yields were determined by gas chromatography and 1H NMR spectroscopy from increases in the GC and 1H NMR peaks due to the corresponding aromatic bromide.Electrochemical measurements
Second harmonic ac voltammetry (SHACV)39,40 measurement of aromatic substrates were carried out with ALS630B electrochemical analyzers in deaerated MeCN containing 0.20 M Bu4N+ClO4− (TBAP) as a supporting electrolyte at 298 K. The carbon working electrode (BAS, surface i.d. 1.6 mm) was polished with BAS polishing alumina suspension and rinsed with acetone before use. The counter electrode was a platinum wire. The measured potentials were recorded with respect to an Ag/AgNO3 (0.01 M) reference electrode. The Eox values (versusAg/AgNO3) are converted into those versusSCE by addition of 0.29 V.41Laser flash photolysis measurements
Measurements of transient absorption spectra in the photochemical reaction of Acr+–Mes with aromatic substrates and/or aqueous HBr were performed according to the following procedure. Typically, an MeCN solution containing Acr+–Mes (1.0 × 10−4 M) and 1,2,4-trimethoxybenzene (1.0 × 10−3 M) was excited by Nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :YAG laser (continuum, SLII-10, 4–6 ns FWHM) at 430 nm. Time dependence of the transient absorption spectra were measured by using a continuous Xe-lamp (150 W) and an In GaAs-PIN photodiode (Hamamatsu 2949) as a probe light and a detector, respectively. The output from the photodiodes and a photomultiplier tube was recorded with a digitizing oscilloscope (Tektronix, TDS3032, 300 MHz). All experiments were performed at 298 K.
:YAG laser (continuum, SLII-10, 4–6 ns FWHM) at 430 nm. Time dependence of the transient absorption spectra were measured by using a continuous Xe-lamp (150 W) and an In GaAs-PIN photodiode (Hamamatsu 2949) as a probe light and a detector, respectively. The output from the photodiodes and a photomultiplier tube was recorded with a digitizing oscilloscope (Tektronix, TDS3032, 300 MHz). All experiments were performed at 298 K.
Time-resolved fluorescence decay measurements
The fluorescence lifetimes of Me2Acr+–Mes and Me2AcrH+ were measured by a Photon Technology International GL-3300 with a Photon Technology International GL-302, nitrogen laser/pumped dye laser system, equipped with a four channel digital delay/pulse generator (Stanford Research System Inc. DG535) and a motor driver (Photon Technology International MD-5020). Excitation wavelength was 430 nm using a toluene solution containing dimethyl-POPOP [1,4-bis(4-methyl-5-phenyl-2-oxazolyl) benzene] (Dojindo, Japan) as a laser dye.Theoretical calculations
Density functional theory (DFT)42 calculations were performed with Gaussian03 (Revision C.02, Gaussian, Inc.).43 The calculations were performed on a 32-processor QuantumCube™ at the B3LYP/6-31G(d) and B3LYP/6-31+G(d,p) level of theory. Graphical outputs of the computational results were generated with the GaussView software program (ver. 3.09) developed by Semichem, Inc.44Acknowledgements
This work was supported by Grants-in-Aid (Nos. 21750146 and 20108010), a Global COE program, “the Global Education and Research Center for Bio-Environmental Chemistry” from the Japan Society of Promotion of Science (JSPS), Iketani Science and Technology Foundation, The Ministry of Education, Science, Technology of Japan and by KOSEF/MEST through WCU project (R31-2008-000-10010-0) from Korea.Notes and references
- (a) March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed., ed. M. B. Smith and J. March, Wiley, New York, 2007, pp. 698–705 Search PubMed; (b) Comprehensive Organic Transformations, 2nd ed., ed. R. C. Larock, Wiley-VCH, New York, 1999, pp. 622–624 Search PubMed.
- (a) B. Das, K. Venkateswarlu, M. Krishnaiah and H. Holla, Tetrahedron Lett., 2006, 47, 8693 CrossRef CAS; (b) B. Das, K. Venkateswarlu, A. Majhi, V. Siddaiah and K. R. Reddy, J. Mol. Catal. A: Chem., 2007, 267, 30 CrossRef CAS.
- A. Podgorsek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8450.
- A. Molinari, G. Varani, E. Polo, S. Vaccari and A. Maldotti, J. Mol. Catal. A: Chem., 2007, 262, 156 CrossRef CAS.
- G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770 CrossRef CAS.
- F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310 CrossRef CAS.
- G. A. Olah, T. Mathew, E. R. Marinez, P. M. Esteves, M. Etzkorn, G. Rasul and G. K. S. Prakash, J. Am. Chem. Soc., 2001, 123, 11556 CrossRef CAS.
- A. Messerschmidt and R. Wever, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 392 CrossRef CAS.
- (a) K. Kikushima, T. Moriuchi and T. Hirao, Chem.–Asia. J., 2009, 4, 1213 Search PubMed; (b) T. Moriuchi, M. Yamaguchi, K. Kikushima and T. Hirao, Tetrahedron Lett., 2007, 48, 2667 CrossRef CAS.
- V. Nair, S. B. Panicker, A. Augstine, T. G. George, S. Thomas and M. Vairamani, Tetrahedron, 2001, 57, 7417 CrossRef CAS.
- L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147 CrossRef CAS.
- K. G. Dewkar, V. S. Narina and A. Sudalai, Org. Lett., 2003, 5, 4501 CrossRef CAS.
- (a) C. Ye and M. J. Shreeve, J. Org. Chem., 2004, 69, 8561 CrossRef CAS; (b) V. A. Joshi, M. Baidossi, Mulhopadhyay and Y. Sasson, Org. Process Res. Dev., 2004, 8, 568 Search PubMed.
- P. K. Chhattise, A. V. Ramaswamy and S. B. Waghmode, Tetrahedron Lett., 2008, 49, 189 CrossRef CAS.
- L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147 CrossRef CAS.
- (a) E. Baciocchi, C. Crescenzi and O. Lanzalunga, Tetrahedron, 1997, 53, 4469 CrossRef CAS; (b) J. Eriksen, C. S. Foote and T. L. Parker, J. Am. Chem. Soc., 1977, 99, 6455 CrossRef CAS; (c) N. Soggiu, H. Cardy, J. L. Habib and J. P. Soumillon, J. Photochem. Photobiol., A, 1999, 124, 1 CrossRef CAS; (d) E. Baciocchi, T. Del Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS; (e) Y. Che, W. Ma, Y. Ren, C. Chen, X. Zhang, J. Zhao and L. Zang, J. Phys. Chem. B, 2005, 109, 8270 CrossRef CAS; (f) S. Lacombe, H. Cardy, M. Simon, A. Khoukh, J. P. Soumillon and M. Ayadim, Photochem. Photobiol. Sci., 2002, 1, 347 RSC; (g) T. Pigot, T. Arbitre, M. Hervé and T. S. Lacombe, Tetrahedron Lett., 2004, 45, 4047 CrossRef CAS; (h) V. Latour, T. Pigot, M. Simon, H. Cardy and S. Lacombe, Photochem. Photobiol. Sci., 2005, 4, 221 RSC.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem.–Eur. J., 2006, 12, 4844 CrossRef CAS.
- N. L. Bauld, in Advances in Electron Transfer Chemistry, ed. P. S. Mariano, JAI Press, Greenwich, CT, 1992, vol. 2, pp. 1–66 Search PubMed.
- (a) F. Müller and J. Mattay, Chem. Rev., 1993, 93, 99 CrossRef; (b) M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550 CrossRef; (c) V. Dichiarante, M. Fagnoni, M. Mella and A. Albini, Chem.–Eur. J., 2006, 12, 3905 CrossRef CAS; (d) E. Baciocci, M. Bietti and O. Lanzalunga, Acc. Chem. Res., 2000, 33, 243 CrossRef CAS; (e) T. Herberts, F. Blume and H. D. Roth, J. Am. Chem. Soc., 1998, 120, 4591 CrossRef CAS.
- (a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS; (b) K. Ohkubo, H. Kotani and S. Fukuzumi, Chem. Commun., 2005, 4520 RSC.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601 RSC.
- (a) H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999 CrossRef CAS; (b) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS; (c) K. Ohkubo, R. Iwata, T. Yanagimoto and S. Fukuzumi, Chem. Commun., 2007, 3139 RSC; (d) K. Ohkubo, T. Nanjo and S. Fukuzumi, Catal. Today, 2006, 117, 356 CrossRef CAS; (e) K. Ohkubo, K. Yukimoto and S. Fukuzumi, Chem. Commun., 2006, 2504 RSC; (f) K. Ohkubo, R. Iwata, S. Miyazaki, T. Kojima and S. Fukuzumi, Org. Lett., 2006, 8, 6079 CrossRef CAS.
- K. T. Potts, M. J. Cipullo, P. Ralli and G. Theodoridis, J. Org. Chem., 1982, 47, 3027 CrossRef CAS.
- (a) P. Arsenyan, E. Paegle and S. Belyakov, Tetrahedron Lett., 2010, 51, 205 CrossRef CAS; (b) P. Arsenyan, O. Pudova, J. Popelis and E. Lukevics, Tetrahedron Lett., 2004, 45, 3109 CrossRef CAS.
- T. J. Dingemans, N. S. Murthy and E. T. Samulski, J. Phys. Chem. B, 2001, 105, 8845 CrossRef CAS.
- M. M. Murray, P. Kaszynski, D. A. Kaisaki, W. Chang and D. A. Dougherty, J. Am. Chem. Soc., 1994, 116, 8152 CrossRef CAS.
- (a) S. Fukuzumi, H. Kotani and K. Ohkubo, Phys. Chem. Chem. Phys., 2008, 10, 5159 RSC; (b) S. Fukuzumi, R. Hanazaki, H. Kotani and K. Ohkubo, J. Am. Chem. Soc., 2010, 132, 11002 CrossRef CAS.
- For formation of π-dimer radical cations of aromatic compounds with near-IR absorption bands, see: (a) J. K. Kochi, R. Rathore and P. Le Magueres, J. Org. Chem., 2000, 65, 6826 CrossRef CAS; (b) S. V. Rosokha, M. D. Newton, A. S. Jalilov and J. K. Kochi, J. Am. Chem. Soc., 2008, 130, 1944 CrossRef CAS; (c) D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850 CrossRef CAS.
- P. K. Das and K. Borowski, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1009 RSC.
- In the presence of 20 mM HBr, no transient absorption spectrum was observed because all of the photoinduced events were completed in microseconds which is time-resolved limitation for our nanosecond laser flash system.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459 CrossRef CAS.
- D. T. Sawyer, T. S. Calderwood, K. Yamaguchi and C. T. Angelis, Inorg. Chem., 1983, 22, 2577 CrossRef CAS.
- No H2O2 was observed after the photocatalytic reaction as checked by iodometric titration and 1H NMR spectroscopy.
- A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron, 2009, 65, 4429 CrossRef CAS.
- P. B. Markel, P. L. Joseph, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2009, 74, 5163 CrossRef.
- K. Ohkubo, H. Kotani, J. Shao, Z. Ou, K. M. Kadish, G. Li, R. K. Pandey, M. Fujitsuka, O. Ito, H. Imahori and S. Fukuzumi, Angew. Chem., Int. Ed., 2004, 43, 853 CrossRef CAS.
- K. Ohkubo, K. Suga, K. Morikawa and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 12850 CrossRef CAS.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518 CrossRef CAS.
- Electrochemical Methods, Fundamental and Applications, ed. A. J. Bard and L. R. Faulkner, John Wiley & Sons, New York, 2001, chap. 10, pp. 368–416 Search PubMed.
- The SHACV method provides a superior approach to directly evaluating the one-electron redox potentials in the presence of a follow-up chemical reaction, relative to the better-known dc and fundamental harmonic ac methods (a) A. M. Bond and D. E. Smith, Anal. Chem., 1974, 46, 1946 CrossRef CAS; (b) E. M. Arnett, K. Amarnath, N. G. Harvey and J.-P. Cheng, J. Am. Chem. Soc., 1990, 112, 344 CrossRef CAS.
- Electrochemical Reactions in Non-aqueous Systems, ed. C. K. Mann and K. K. Barnes, Mercel Dekker, New York, 1970 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) W. J. Hehre, L. Radom, P.v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- Gaussian 03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- R. Dennington II, T. Keith, J. Millam, K. Eppinnett, W. L. Hovell and R. Gilliland, Semichem, Inc., Shawnee Mission, KS, 2003.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR chemical shift of products (S1) and time course in bromination of TMB. See DOI: 10.1039/c0sc00535e |
| This journal is © The Royal Society of Chemistry 2011 |