Molecular anchors in the solid state: Restriction of intramolecular rotation boosts emission efficiency of luminogen aggregates to unity†
Zujin
Zhao
a,
Ping
Lu
b,
Jacky W. Y.
Lam
a,
Zhiming
Wang
b,
Carrie Y. K.
Chan
a,
Herman H. Y.
Sung
a,
Ian D.
Williams
a,
Yuguang
Ma
b and
Ben Zhong
Tang
*ac
aDepartment of Chemistry and Institute of Molecular Functional Materials, The Hong Kong University of Science & Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
bState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun, 130012, China
cDepartment of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China
First published on 24th December 2010
Abstract
Introduction of freely rotatable tetraphenylethene (TPE) to conventional luminophors quenches their light emissions in the solutions but endows the resultant molecules (TPEArs) with aggregation-induced emission characteristics in the condensed phase due to the restriction of intramolecular rotation. High fluorescence quantum yields up to 100% have been achieved in the films of TPEArs.
Introduction
Synthesis of luminescent materials with efficient light emissions in the solid state is a hot research topic. One problem associated with the dye emission is aggregation-caused quenching (ACQ): in poor solvents or during film formation, the dye molecules aggregate, which often quenches their light emissions due to the formation of detrimental species such as excimers and exciplexes.1 This notorious ACQ effect has prevented many lead luminogens identified by the laboratory solution-screening process from finding real-world applications. To mitigate the ACQ problem, various chemical, physical, and engineering approaches and processes have been developed. The attempts have, however, met with only limited success. The difficulty lies in the fact that aggregate formation is an intrinsic process when luminogenic molecules are located in close vicinity in the condensed phase. It will be nice if a system can be developed, in which light emission is enhanced, rather than quenched, by aggregation. In 2001, we found such a system and observed a novel phenomenon of aggregation-induced emission (AIE):2,3 a series of propeller-like, nonemissive molecules such as silole and tetraphenylethene (TPE) are induced to emit intensely by aggregate formation.Through a series of designed experiments and theoretical calculations, we identified restriction of intramolecular rotation (IMR) as the main cause for the AIE effect.4 In the solution state, the active rotation of the phenyl blades around the dye stator effectively deactives the excited state via rotational energy relaxation channels, thus rendering the dyes nonemissive. In the aggregate state, the IMR process is impeded, which blocks the non-radiative decay pathways and hence converts the dyes into strong emitters. Traditional luminophors are usually flat disk-like aromatic molecules and experience severe ACQ effect due to strong π–π intermolecular interactions in concentrated solutions and aggregate and crystal states. We envisioned that introduction of twisted AIE molecules as substituents to these luminophors will disrupt their planarity and hence may solve their ACQ problem. Meanwhile, new AIE luminogens with new and/or enhanced optical properties may be generated through such strategy. With such regard, in this paper, we functionalized TPE to a series of “conventional” planar luminophores such as pyrene and anthracene and presented the emission behaviors of the resultant molecules.
Results and discussion
The synthetic routes to the TPE-substituted luminophors (TPEArs) are illustrated in Scheme 1. The detailed synthetic procedures and characterization data are given in the Electronic Supplementary Information (ESI).† All the desirable products are obtained from moderate to high yields. Single crystals of TPEArs are grown from their methanol–dichloromethane solutions and analyzed by X-ray diffraction crystallography. The crystal structures of TPEArs are shown in Fig. 1 and their crystal analysis data are given in Tables S1 and S2 in the ESI.† All the dye molecules are soluble in common organic solvents such as THF, but insoluble in water.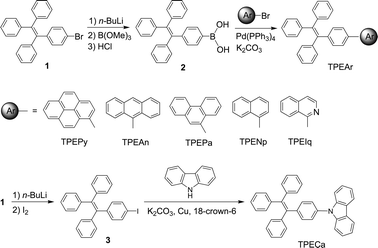 | ||
| Scheme 1 Synthetic routes to the TPE-substituted planar luminophors (TPEArs). | ||
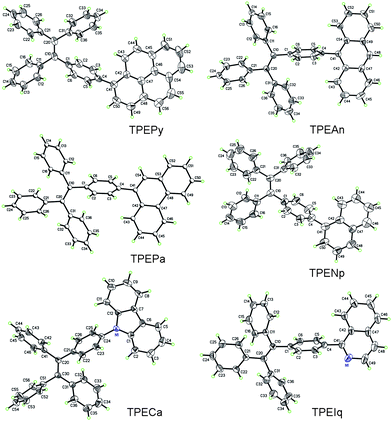 | ||
| Fig. 1 ORTEP drawings of TPEArs. | ||
Fig. 2a shows the absorption spectra of TPEArs in THF solutions. The spectral profile and peak absorptivity vary largely with the type of planar luminogenic unit. TPEAn and TPEPy show redder absorptions at 387 and 348 nm, corresponding to the π–π* transitions of the anthracene and pyrene units, respectively. The absorption maxima of other molecules are located at 321–337 nm. Upon photoexcitation, the dilute THF solutions (10 μM) of TPEPy and TPEAn show weak photoluminescence (PL) peaked at ∼432 and ∼423 nm, respectively. Under the same measurement conditions, only noisy PL signals without discernable peaks are recorded in other TPEArs, revealing that they are practically nonluminescent when molecularly dissolved in good solvents. The fluorescence quantum yields (ΦF's) of TPEAn and TPEPy are measured to be 0.28 and 0.34%, respectively, which are much lower than those of anthracene (36%), pyrene (32%) and their derivatives.5–7 The ΦF values of other TPEArs are even lower and fall in the range of 0.019–0.045% (Table 1).
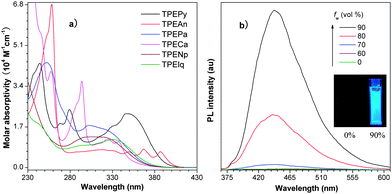 | ||
| Fig. 2 a) Absorption spectra of TPEArs in THF solutions. b) PL spectra of TPEPy in THF–water mixtures with different water contents (fw). Insert in b): photographs of TPEPy in THF–water mixtures (left, fw = 0; right, fw = 90%) taken under UV illumination. Excitation wavelength: 350 nm. | ||
| λ abs (nm) | λ em (nm) | ΦF (%) | ||||
|---|---|---|---|---|---|---|
| Soln | Soln | Cryst | Film | Solnd | Filme | |
| a In THF (10 μM) solution. b Grown from methanol–dichloromethane mixture. c Film drop-casted on quartz plates. d Quantum yields (ΦF) determined in THF using 9,10-diphenylanthracene (ΦF = 90% in cyclohexane) as standard. e Quantum yields of the films measured by integrating sphere. f For its pyrene parent, ΦF = 32% in solution. g For its anthracene parent, ΦF = 36% in solution. | ||||||
| TPEPy f | 348 | 432 | 443 | 468 | 0.34 | 100 |
| TPEAn g | 387 | 423 | 428 | 450 | 0.28 | 100 |
| TPEPa | 323 | 444 | 481 | 0.033 | 88 | |
| TPENp | 321 | 452 | 469 | 0.022 | 83 | |
| TPECa | 337 | 440 | 468 | 0.045 | 100 | |
| TPEIq | 331 | 445 | 471 | 0.019 | 20 | |
All the TPEArs are less emissive than their corresponding planar luminogenic units in the solution state, suggesting that the TPE moiety works as a PL quencher. This is somewhat surprising but understandable when we take the IMR process of TPE into consideration. The multiple phenyl blades of the TPE unit in an isolated molecule of TPEArs can undergo active IMR process with little restraint in the dilute solutions. Collectively, these multiple molecular motions quickly consume the photonic energy of the excited state. The swift dissipation of the photonic energy as thermal energy effectively deactivates the excitons of TPEArs and thus quenches their light emissions in the solutions.
Similar to TPE, the dye molecules become strong emitters when aggregated. As shown by the example in Fig. 2b, the emission of TPEPy is intensified when a large amount of water (fw >70%) is added into its THF solution. The higher the water content, the stronger is the light emission. Since water is a non-solvent for TPEPy, its molecules must have aggregated in the aqueous mixtures with high water contents. Clearly, the PL of TPEPy is enhanced by aggregate formation. Higher water content populates the aggregates, thereby boosting its light emission to a greater extent. Similar emission enhancement behaviors are also observed in other TPEArs, suggesting that the attachment of the TPE unit to “conventional” luminophors has endowed the resultant molecules with a novel feature of AIE.
Like their aggregates suspended in the aqueous media, TPEArs emit intensely in the solid state. UV irradiation of their crystals gives deep blue PL's with maxima at 428–452 nm (Fig. 3a). The crystal emissions of TPEPy and TPEAn are found at wavelengths close to those in the THF solutions. This suggests the PL's are originated from the same radiative decay of singlet excitons induced by photoexcitation. The thin films of TPEArs are also highly emissive but their PL spectra are broader and observed at longer wavelengths (Fig. 3b). The ΦF's of their films are much higher than the solution values. The ΦF values measured by integrating sphere are 100% in TPECa, TPEAn, and TPEPy, which are much higher than those of pyrene, anthracene, and even TPE (49.2%),8 thanks to the synergistic electronic interactions between the planar and twisted luminogenic units.
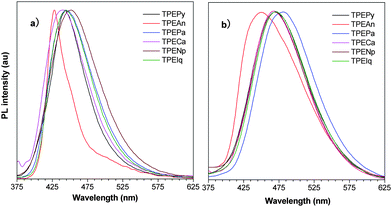 | ||
| Fig. 3 PL spectra of a) crystals and b) films of TPEArs. Excitation wavelength: 350 nm. | ||
Crystallization generally red-shifts emission and decreases emission efficiency. Why are opposite behaviors observed in TPEArs? Similar to other AIE molecules,2e,f,g during the crystallization process, the molecules of TPEArs may have conformationally adjusted themselves by twisting their phenyl rings to fit into the crystalline lattices. The crystal data show that all the molecules adopt highly twisted conformations in the crystal state due to the propeller-shaped TPE unit. The torsion angles between the planar luminophors and the directly linked phenyl rings of the TPE units are 66.74° (TPEPy), 75.27° (TPEAn), 58.10° (TPEPa), 78.85° (TPECa), 51.76° (TPENp), and 52.73° (TPEIq). TPEAn and TPECa exhibit the highest torsion angles because of the severe steric hindrance between the TPE moieties and the big, flat anthracene and carbazole rings. The conformations of the molecules strongly affect their HOMO and LUMO energy levels. The calculated molecular orbitals of TPENp, TPEPa, TPEPy, and TPEAn are displayed in Fig. 4, and those of TPEIq and TPECa are given in Table S3 in the ESI.† The HOMO and LUMO of TPEPa and TPENp are dominated by the orbitals from the TPE and planar aromatic rings, revealing that their PL's stem from the exciton decay of the whole molecules. However, the large torsion angles between the two chromophores in TPEPy and TPEAn lead to little orbital overlapping and poorer electronic communication. Consequently, in these molecules, the TPE unit contributes less to the energy levels and the electron densities are mainly located on the pyrene and anthracene rings. Such electron distribution manifests that the absorption and emission of the molecules are mainly controlled by the planar chromophores.9 This may also explain why TPEAn and TPEPy are still somewhat emissive in the solutions because the IMR process of the TPE unit is not working directly on the PL process of the whole molecules.2e,g
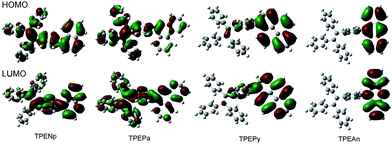 | ||
| Fig. 4 Molecular orbital amplitude plots of HOMO and LUMO levels of TPENp, TPEPa, TPEPy and TPEAn calculated using the B3LYP/6-31G(d) basis set. | ||
In an effort to further understand the mechanism operating in this AIE system, we checked the geometries and packing arrangements of TPEArs in the crystal state. The packing models of crystals of TPEPy, TPEAn, TPEPa, and TPECa are resembled to anchors (Fig. 5). The planar aromatic rings are situated between two TPE units, which efficiently hampers their π–π interactions and hence excimer formation. The TPE units are also sandwiched between two planar units. Multiple C–H⋯π hydrogen bonds with distances of 2.719–3.090 Å are formed between the hydrogen atoms of the phenyl rings of the TPE unit in one molecule and the π cloud of large planar aromatic ring in another molecule. These multiple C–H⋯π hydrogen bonds help rigidify the molecular conformation and lock the molecular rotation. As a result, the excited state energy consumed by the IMR process is greatly reduced, which enables the molecules to emit intensely in the solid state. Without such constraint, the TPEAr molecules may assume a more planar conformation in the solid thin films. This enhances the π–π stacking interactions of the planar luminogenic units and hence leads to red-shift and broadening of the PL spectra.
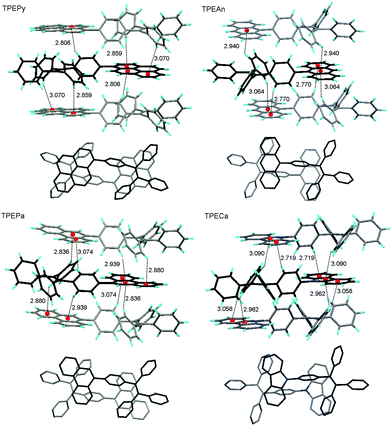 | ||
| Fig. 5 (Upper panel) C–H⋯π hydrogen bonds with indicated distances (Å) between TPEAr adjacent molecules. (Lower panel) Top view of the adjacent TPEAr molecules. | ||
Conclusions
In summary, we have synthesized and investigated the photophysical properties of a series of TPE-substituted luminophors. All the TPEArs are weakly emissive in the solutions due to the IMR process of the TPE unit but are induced to emit intensely in the condensed phase with ΦF values up to unity. Restriction of intramolecular rotation is responsible for such novel AIE effect. The present work not only verifies the mechanism of the AIE phenomenon but also generates promising luminsecent materials for applications in optics and electronics. It also provides a versatile strategy for the creation of efficient solid emitters,10 which takes the advantage of aggregate formation but causes no severe side effects. The construction of efficient light-emitting diodes using these luminogens are currently under investigation in our laboratory and will be reported in a separate paper.Acknowledgements
This project was partially supported by the Research Grants Council of Hong Kong (604509, 603008, 601608, and ITP/008/09NP), the University Grants Committee of Hong Kong (AoE/P-03/08), and the National Science Foundation of China (20634020 and 20974028). B.Z.T. thanks the support from Cao Gaungbiao Foundation of Zhejiang University.Notes and references
- (a) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS; (b) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS; (c) M. Shimizu, H. Tatsumi, K. Mochida, K. Shimono and T. Hiyama, Chem.–Asian J., 2009, 4, 1289 CrossRef CAS; (d) K. Y. Pu and B. Liu, Adv. Funct. Mater., 2009, 19, 277 CrossRef CAS; (e) A. Iida and S. Yamaguchi, Chem. Commun., 2009, 3002 RSC; (f) T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS; (g) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799 CrossRef CAS; (h) S. Yin, Q. Peng, Z. Shuai, W. Fang, Y. Wang and Y. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205409 CrossRef; (i) Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef; (j) A. B. Koren, M. D. Curtis, A. H. Francis and J. W. Kampf, J. Am. Chem. Soc., 2003, 125, 5040 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. A. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70 RSC; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (d) Z. Zhao, Z. Wang, P. Lu, C. Y. K. Chan, D. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2009, 48, 7608 CrossRef CAS; (e) Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef; (f) Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, Y. Dong, H. H. H. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40 RSC; (g) Z. Zhao, S. Chen, X. Shen, F. Mahtab, Y. Yu, P. Lu, J. W. Y. Lam, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 686 RSC.
- (a) M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259 CrossRef CAS; (b) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS; (c) C. Belton, D. F. O'Brien, W. J. Blau, A. J. Cadby, P. A. Lane, D. D. C. Bradley, H. J. Byrne, R. Stockmann and H.-H. Hörhold, Appl. Phys. Lett., 2001, 78, 1059 CrossRef CAS; (d) L. Antolini, E. Tedesco, G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, D. Casarini, G. Gigli and R. Cingolani, J. Am. Chem. Soc., 2000, 122, 9006 CrossRef CAS; (e) R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565 CrossRef CAS.
- (a) Z. Li, Y. Dong, B. Mi, Y. Tang, M. Häussler, H. Tong, P. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sun, K. Wong, P. Gao, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061 CrossRef CAS; (b) G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Z. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS.
- (a) J. N. Moorthy, P. Venkatakrishnan, P. Natarajan, D.-F. Huang and T. J. Chow, J. Am. Chem. Soc., 2008, 130, 17320 CrossRef CAS; (b) M. Takahashi, T. Odagi, H. Tomita, T. Oshikawa and Mi. Yamashita, Tetrahedron Lett., 2003, 44, 2455 CrossRef CAS; (c) Z. Zhao, S. Yu, L. Xu, H. Wang and P. Lu, Tetrahedron, 2007, 63, 7809 CrossRef CAS; (d) M.-T. Lee, C.-H. Liao, C.-H. Tsai and C. H. Chen, Adv. Mater., 2005, 17, 2493 CrossRef CAS; (e) A. Iida and S. Yamaguchi, Chem. Commun., 2009, 3002 RSC.
- (a) K.-C. Wu, P.-J. Ku, C.-S. Lin, H.-T. Shih, F.-I. Wu, M.-J. Huang, J.-J. Lin, I.-C. Chen and C.-H. Cheng, Adv. Funct. Mater., 2008, 18, 67 CrossRef CAS; (b) S. T. Tao, Z. K. Peng, X. H. Zhang, P. F. Wang, C.-S. Lee and S.-T. Lee, Adv. Funct. Mater., 2005, 15, 1716 CrossRef CAS; (c) C. Tang, F. Liu, Y.-J. Xia, L.-H. Xie, A. Wei, S.-B. Li, Q.-L. Fan and W. Huang, J. Mater. Chem., 2006, 16, 4074 RSC; (d) M. Y. Lo, C. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808 CrossRef CAS; (e) Z. Zhao, J.-H. Li, P. Lu and Y. Yang, Adv. Funct. Mater., 2007, 17, 2203 CrossRef CAS.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York and London 1971 Search PubMed.
- (a) R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961 CrossRef CAS; (b) Z. Zhao, S. Chen, J. W. Y. Lam, C. K. W. Jim, C. Y. K. Chan, Z. Wang, P. Lu, C. Deng, H. S. Kwok, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 7963 CrossRef CAS.
- (a) Y.-H. Kim, H.-C. Jeong, S.-H. Kim, K. Yang and S.-K. Kwon, Adv. Funct. Mater., 2005, 15, 1799 CrossRef CAS; (b) S.-K. Kim, Y.-I. Park, I.-N. Kang and J.-W. Park, J. Mater. Chem., 2007, 17, 4670 RSC.
- (a) Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221 RSC; (b) W. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for the synthesis of TPEArs, crystal data of TPEArs, and calculated molecular orbitals of TPEIq and TPECa. CCDC reference numbers 753334–753339. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00521e |
| This journal is © The Royal Society of Chemistry 2011 |