A fully self-assembled non-symmetric triad for photoinduced charge separation†
Elisabetta
Iengo
*ab,
G. Dan
Pantoş
a,
Jeremy K. M.
Sanders
a,
Michele
Orlandi
c,
Claudio
Chiorboli
d,
Sandro
Fracasso
c and
Franco
Scandola
*ce
aUniversity of Cambridge, Chemical Laboratories, Lensfield Road, Cambridge, CB2 1EW, UK
bDipartimento di Scienze Chimiche e Farmaceutiche, Università degli Studi di Trieste, Via L. Giorgieri 1, 34127, Trieste, Italy. E-mail: eiengo@units.it; Fax: +39 040-558-3903; Tel: +39 040-558-3955
cDipartimento di Chimica, Università di Ferrara, Via L. Borsari 46, 44100, Ferrara, Italy. E-mail: snf@unife.it; Fax: +39 0532-240709; Tel: +39 0532-455160
dISOF-CNR Sezione di Ferrara, Via L. Borsari 40, 44100, Ferrara, Italy
eCentro Interuniversitario di Ricerche sulla Conversione Chimica dell'Energia Solare, Sezione di Ferrara, Via L. Borsari 46, 44100, Ferrara, Italy
First published on 24th December 2010
Abstract
A very efficient and successful metal-mediated strategy towards the formation of a non-symmetric triad is described: appropriate Lewis acid and/or base functions on the molecular components (a naphthalenediimide, an aluminium(III) porphyrin, and a ruthenium(II) porphyrin) lead to the desired product uniquely. The photophysics of the triad was investigated in detail using time-resolved spectroscopy in the pico- and nanosecond time domains. The strategy is of great potential interest as, while confining the synthetic effort to the single components, it can give access to a wide range of photoactive systems.
Introduction
Artificial photosynthesis, i.e., the conversion of light energy into fuels by artificial systems, is an active research field, with interesting potential for a long-term solution to the world demand for clean energy.1 In recent years, following a biomimetic approach, a variety of chemical systems aimed at reproducing the behaviour of the most important functional units of natural photosynthesis, e.g., multichromophoric antenna systems,2–8 charge-separating reaction centers,9–13 and multielectron redox catalysts,14–20 have been designed, synthesized, and functionally characterized. As the functions generally rely on the occurrence of very specific sequences of intercomponent energy or electron transfer elementary steps, a very precise spatial organization of molecular components (chromophores, electron donors and acceptors) must be synthetically achieved. To this end, classical organic synthesis has been widely used in the construction of functional units for artificial photosynthesis.Despite its advantages from the viewpoint of robustness and structural definition, the covalent approach is demanding both in terms of synthesis and of molecular design. Perhaps at the expense of some structural rigidity, softer, non-covalent strategies are advantageous in terms of mild reaction conditions, combinatorial flexibility, and repair capabilities. Among these, considerable interest has been given to the use of metal coordination for assembling molecular components into appropriate functional structures (“metal-mediated” approach).21 In this approach, depending on the type of metal used and of ligand function present on the molecular component, the assembling process may run under kinetic control (chemical synthesis, relatively mild conditions) or under thermodynamic control (true self-assembling).
In the last few years we have exploited the exocyclic coordination ability of meso-substituted pyridyl porphyrins to produce 2 + 2 porphyrin/metal complex molecular squares,22 side-to-face multiporphyrin arrays,23 and more complex structures combining these two structural motifs.24 Such systems behave as antenna systems, characterized by efficient energy transfer processes between the various porphyrin units, with energy gradients and spin multiplicity of the processes determined by synthetic design. Related systems based on pyridyl-substituted aromatic dicarboxyimides have been produced, which undergo additional photoinduced electron transfer processes.25 The Sanders group has produced a number of self-assembled heterometallic oligoporphyrins, exploiting the different coordination behaviours of Zn(II)-, Ru(II)-, Rh(III)-, and Sn(IV)-porphyrins.26 They recently reported on the efficient use of aluminium porphyrins as building units for the formation of multi-porphyrin arrays (either discrete or polymeric).27 During the present investigation, we have inserted an aluminium centre in a pyridyl porphyrin, and we demonstrate the versatility of such multi-site (both donor and bis-acceptor) equipped units.
The metal-mediated self-assembling strategies developed until now are ideally suited to the construction of symmetric multichromophoric systems. It must be pointed out, however, that some functional units of interest to artificial photosynthesis, notably triads and more complex systems for photoinduced charge separation, are intrinsically non-symmetric. Implementation of metal-mediated self-assembling to the construction of triad systems is much less obvious, as it requires molecular recognition between the subunits to be assembled (Fig. 1). This can only be achieved if highly selective metal–ligand interactions are used, and different types of such interactions are simultaneously implemented. In this work, a first attempt28–30 towards the challenging objective of a self-assembling triad is presented.
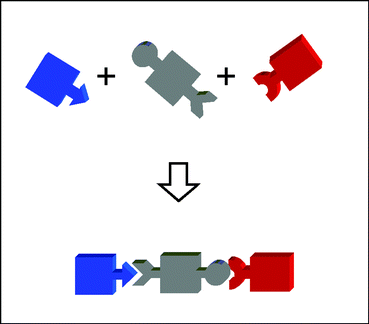 | ||
| Fig. 1 Schematic representation of self-assembling of a non-symmetric triad. | ||
The molecular building units chosen are: (i) an aluminium porphyrin as the central photoexcitable chromophore (ii) a naphthalenediimide as electron acceptor unit, and (iii) a ruthenium porphyrin as electron donor unit. The selective affinity of the two different metallo-porphyrins towards nitrogen or oxygen ligands is crucial for the efficient self-sorting of the desired three-component products. In particular, Al(III)-porphyrins can axially bind a carboxylic group (and with a much weaker affinity a nitrogen based ligand),27 whereas Ru(II)(CO)-porphyrins can axially bind a pyridyl group.31 Thus, efficient assembling is implemented by introduction of a carboxylic function on the naphthalenediimide unit and of a pyridyl appended ligand on the aluminium porphyrin. As will be shown in the following sections, a single three-component adduct, NDI–AlMPyP–RuP, quantitatively self-assembles from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of the three starting units (Scheme 1).
:1:1 mixture of the three starting units (Scheme 1).
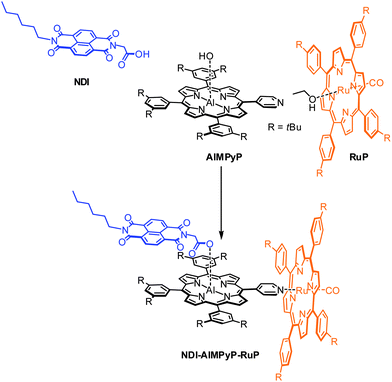 | ||
| Scheme 1 Self-assembling of the non-symmetric triad NDI–AlMPyP–RuP. | ||
Two related dyads, NDI–AlP and RuP–NDI′′–RuP,32 have also been designed for purposes of comparison (Chart 1).
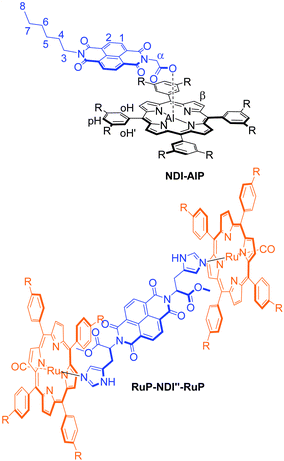 | ||
| Chart 1 Dyad systems designed for comparison studies. | ||
The assembling, and photophysical behaviour of these systems has been investigated in solution and discussed by comparison with appropriate models of the individual molecular components (NDI′, AlP(ba), RuP(py), AlMPyP(ba), and AlMPyP–RuP (Chart 2)).
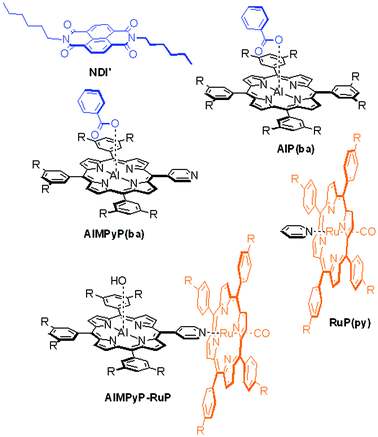 | ||
| Chart 2 Model compounds used for comparison studies. | ||
Results and discussion
A complete and detailed Experimental Section is reported in the ESI.† All the systems reported have been characterized in solution by means of conventional 1D and 2D NMR experiments.‡,§Assembly of NDI–AlP and RuP–NDI′′–RuP (Chart 1) is straightforward. These dyads are obtained quantitatively by simple mixing of the components, in the correct ratios, in chloroform solution at room temperature. NDI axially binds to the aluminium porphyrin, AlP, via the carboxylate with elimination of water, whereas NDI′′ connects to two ruthenium porphyrins, RuP, via the imidazoles. In Fig. 2 the 1H NMR spectrum (CDCl3) of NDI–AlP, compared to the spectrum (DMSO-d6) of free NDI, is reported.33 Relative integration of the resonances of NDI and AlP confirms the formation of a 1:1 adduct; the signals of NDI, corresponding to the protons closer to the metal centre, are shifted upfield as expected for an axially connected species (e.g., Δδ = −3.37 for α). All the resonances are sharp, indicating the formation of a stable adduct.
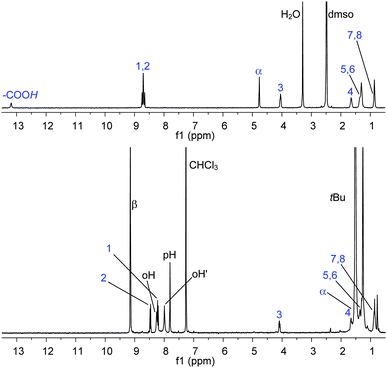 | ||
| Fig. 2 1H NMR (CDCl3), bottom, of NDI–AlP, compared to the 1H NMR (DMSO-d6), top, of NDI. See Chart 1 for labelling scheme. Assignments have been made with 2D NMR experiments (COSY and HSQC). | ||
The 1H NMR solution characterization of RuP–NDI′′–RuP is reported in the ESI (Figure SI5, SI6).† The axial coordination of the bisimide, NDI′′ to two metallo-porphyrins, RuP, is confirmed by relative integration and by the strong upfield shift of the resonances of the central bisimide unit (e.g., Δδ = −5.66 for 2 and Δδ = −5.85 for 3 of the imidazole ring, see also Figure SI3 and SI5, ESI†).
The characterization of AlMPyP is less straightforward. Insertion of aluminium in the monopyridyl porphyrin core proceeds smoothly, as already reported for its tetraphenyl porphyrin analogue.27a This porphyrin self-assembles in chloroform solution to give oligomeric structures of the type (AlMPyP)n, as the pyridyl group of one molecule is axially bound to the 6th coordination position of the aluminium of another molecule (Scheme 2).
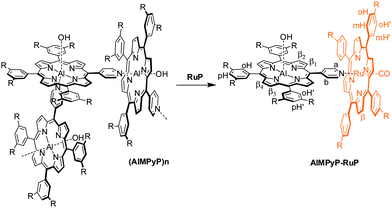 | ||
| Scheme 2 Schematic representation of the self-assembled species (AlMPyP)n and of the formation of the side-to-face adduct AlMPyP–RuP, upon addition of RuP to (AlMPyP)n. | ||
This type of interaction is not very strong,34 probably leading to the presence of several oligomeric assemblies in equilibrium, similar to that already observed for zinc pyridyl porphyrins.35 As a consequence the 1H NMR spectrum (CDCl3) of AlMPyP shows broad, concentration dependent, resonances (Fig. 3).
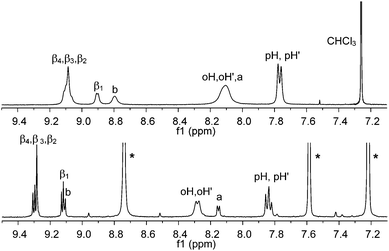 | ||
| Fig. 3 Aromatic region of the 1H NMR of AlMPyP in CDCl3, top, and pyridine-d5, bottom. See Scheme 2 for labelling scheme. Assignments have been made with a 2D H–H COSY experiment. Peaks of residual non-deuterated pyridine are indicated with an asterisk. | ||
In particular, broad and partly upfield shifted signals result from the pyridyl (a and b), β1 and β2protons as they fall in the shielding region of the porphyrin ring current in these self-assembled species (Fig. 3, top). Addition of an excess of competing pyridine-d5 gives rise to a sharp well-resolved 1H NMR spectrum (Fig. 3, bottom).36
Analogously, addition of equimolar amounts of RuP breaks the weak aluminium–pyridyl bond in favour of the more stable and inert ruthenium–pyridyl interaction, thus leading to the selective and quantitative formation of AlMPyP–RuP. This side-to-face bis metallo-porphyrin assembly, obtained at room temperature in a few minutes, shows only sharp resonances in the 1H NMR spectrum (Fig. 4), with the typical upfield shifts for the resonances of the side metallo-porphyrin AlMPyP (e.g., Δδ = −7.19, −2.13 and −1.63 for a, b and β1 of AlMPyP, respectively). In particular the ruthenium porphyrin acts as a shift reagent for the β protons of AlMPyP which resonances are well resolved and spread over ca. 2 ppm. The phenyl protons of both the metallo-porphyrins are split into two resonances each, as for both units the two sides of the porphyrin planes are not equivalent. The rotation along the Cmeso–Cring bond is slow on the NMR timescale at room temperature for RuP and intermediate-to-fast for AlMPyP, resulting in distinct sharp resonances for the ortho and metaprotons of RuP and distinct but broad resonances for the orthoprotons of AlMPyP.
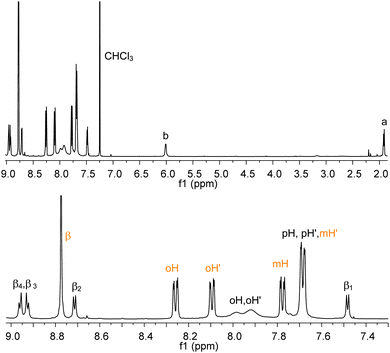 | ||
| Fig. 4 1H NMR (CDCl3) of AlMPyP–RuP (top, tert-butyl groups omitted for clarity) and its enlarged aromatic region (bottom). See Scheme 2 for labelling scheme. Assignments have been made with a 2D H–H COSY experiment. | ||
The target triad, NDI–AlMPyP–RuP, can be easily obtained by subsequent addition of an equimolar amount of NDI to AlMPyP–RuP (Scheme 3).
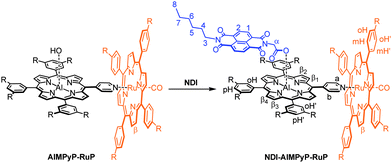 | ||
| Scheme 3 Formation of the non-symmetric triad NDI–AlMPyP–RuP by addition of a stoichiometric amount of NDI to AlMPyP–RuP. | ||
The bisimide binds axially to the aluminium centre (with elimination of water) via the carboxylic functionality. The reaction is selective, quantitative and takes place at room temperature (stirring is necessary to favour the dissolution of NDI, which is barely soluble in chloroform by itself). In Fig. 5 the 1H NMR spectrum (CDCl3) of NDI–AlMPyP–RuP is shown.
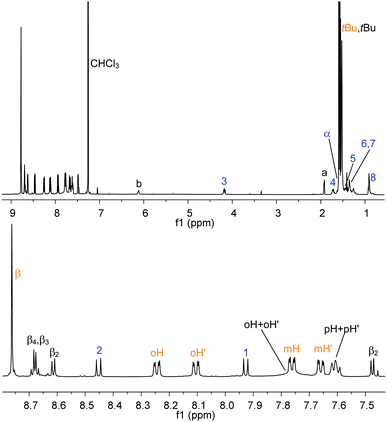 | ||
| Fig. 5 1H NMR (CDCl3) of NDI–AlMPyP–RuP (top) and its enlarged aromatic region (bottom). See Scheme 2 and Scheme 3 for labelling scheme. Assignments have been made using 2D NMR experiments (COSY, HSQC, HMBC, NOESY). | ||
Most of the protons show well-resolved and sharp signals, reflecting the stability and absence of scrambling processes. The two-axial coordination motif of the assembly (i.e., NDI axially connected to the aluminium porphyrin and the aluminium porphyrin axially connected to the ruthenium one), is evident from the shift of the resonances pertaining to the protons closer to the shielding cones of the porphyrin macrocycles. Not surprisingly, the 1H NMR of NDI–AlMPyP–RuP, in terms of chemical shifts, is close to the overlap of the 1H NMR spectrum of AlMPyP–RuP and of the NDI part of the 1H NMR of NDI–AlP (e.g., Δδ = −3.38 for α, Δδ = −2.03 for b and Δδ = −7.20 for a). Importantly, the same final three-component assembly, NDI–AlMPyP–RuP, forms uniquely either by sequential addition of the starting units, in any order or, more strikingly, by mixing the three units in stoichiometric ratio one-pot (Scheme 1). Therefore, the affinity of the metal centers, together with the metal coordination number and the appropriate functionalization of the modules, is fundamental for the success of the self-selection process and the production of a single stable product.
Stability in solution, spectroscopic and electrochemical properties
As already observed for related side-to-face and axially coordinated assemblies,23,36 the dyad and triad systems of this work are truly supramolecular in nature. For NDI–AlP and NDI–AlMPyP–RuP this is demonstrated by the appreciable additivity of the absorption spectra of the subunits (Fig. 6).38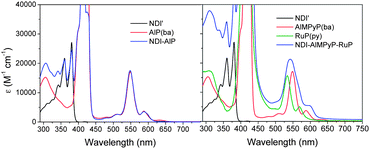 | ||
| Fig. 6 Absorption spectra of NDI–AlP (left) and NDI–AlMPyP–RuP (right) and of their model compounds. | ||
On close inspection, however, some minor differences between the spectrum of the NDI–AlMPyP–RuP triad and the sum of those of the molecular components can be noticed, in particular in a red shift of the lowest energy aluminium porphyrin band at ca. 600 nm. This shift, very similar to that observed when AlMPyP(ba) self-associates in concentrated solution,37 tends to disappear upon strong dilution (<5 × 10−5 M) of the solutions and can be taken as a marker of the coordination of the pyridyl group. These spectrophotometric dilution experiments (monitoring the stability of the AlMPyP–RuP linkage), together with spectrofluorimetric dilution experiments on the NDI–AlP dyad (vide infra, monitoring the stability of the NDI–AlP linkage), lead to the conclusion that the dyad and triad systems are stable in dichloromethane solution down to a concentration of 5 × 10−5 M.
The supramolecular nature is also apparent when the electrochemistry of these systems is compared to that of the component subunits (Table 1). For example, in the NDI–AlMPyP–RuP triad, the reduction of the aluminium porphyrin, the two reductions of the NDI unit, the oxidation of the ruthenium porphyrin and the oxidation of the aluminium porphyrin can be easily assigned, as their potentials are identical within experimental error with those of the constituent units.
| Reduction | Oxidation | ||||
|---|---|---|---|---|---|
| a In CH2Cl2 at 298 K, 0.1 M TBA(PF6) as electrolyte, SCE as reference electrode, ferrocene (0.46 V vs.SCE39) as internal standard. b Concentration independent in the 1 × 10−4–5 × 10−5 M range.40 | |||||
| NDI –AlP | −1.30 | −1.09 | −0.69 | 0.83 | |
| NDI – AlMPyP – RuP | −1.19 | −1.05 | −0.68 | 0.76 | 0.90 |
| RuP(py) | 0.78 | ||||
| AlP(ba) | −1.31 | 0.82 | |||
| AlMPyP(ba) b | −1.21 | 0.88 | |||
| NDI ′ | −1.06 | −0.65 | |||
Photophysics and spectroelectrochemistry of molecular components
The NDI′ model exhibits in dichloromethane the weak structured fluorescence (λmax, 380 nm; τ, ca. 15 ps) typical of this type of chromophore.41At room temperature a dichloromethane solution of AlP(ba) shows a typical metal porphyrin fluorescence, with prominent vibronic bands at 596 and 648 nm and a lifetime of 5.5 ns. At 77 K, in addition to fluorescence, a weak phosphorescent emission can be observed at 782 nm. The spectroscopic signature of the singlet excited state, as obtained by femtosecond absorption spectroscopy (excitation at 550 nm), is shown in Fig. 7a. It is characterized by Q-band bleach at 550 nm (partially covered by the excitation pulse), absorption at longer wavelengths, with superimposed stimulated emission (apparent bleach at ca. 600 and 650 nm). The spectrum is appreciably constant in the 1–1000 ps time scale. The broad excited-state difference absorption spectrum of the triplet state (T1), as obtained by nanosecond laser photolysis (excitation at 532 nm), is shown in Fig. 7b. It decays with oxygen-dependent lifetime (310 μs in deaerated dichloromethane).
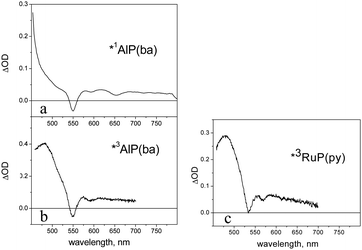 | ||
| Fig. 7 (a) Transient absorption spectrum of the singlet excited state of AlP(ba), obtained by femtosecond spectroscopy (for details, see text); (b) transient absorption spectrum of the triplet excited state of AlP(ba), obtained by nanosecond laser photolysis (for details, see text); (c) transient absorption spectrum of the triplet excited state of RuP(py), obtained by nanosecond laser photolysis. | ||
As it is usual for ruthenium porphyrins,23 the S1 state of the RuP(py) model does not fluoresce but rather undergoes very fast intersystem crossing. The T1 state can be detected at 77 K by a weak phosphorescence (λmax = 726 nm) or at room temperature by nanosecond transient absorption measurements (spectrum in Fig. 7c, lifetime ca. 30 μs in deaerated dichloromethane). Femtosecond spectroscopy shows that triplet formation is complete in a sub-picosecond time scale.
The difference spectra obtained in spectroelectrochemistry for one-electron reduction of NDI′ and oxidation of AlP(ba) and RuP(py)42 are shown in Fig. 8. The main spectroscopic signatures for NDI′− are two sharp maxima at 475 and 510 nm. For the AlP(ba)+ and RuP(py)+ radical cations, the most distinctive features are the bleaches of the main Q-band, at 550 and 530 nm, respectively. Sum spectra are also shown in Fig. 8, to simulate the spectral behaviour expected for charge-separated states that may be formed in the dyad and triad systems.
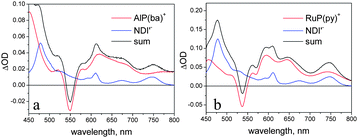 | ||
| Fig. 8 Sum spectra of the one-electron reduced form of NDI′ and the one-electron oxidized forms of AlP(ba) (a) and RuP(py) (b). | ||
Photophysics of the NDI–AlP dyad
A simplified energy level diagram for the NDI–AlP dyad in dichloromethane (high-lying excited states of the NDI unit, omitted for clarity) is shown in Fig. 9a. This diagram is based on the spectroscopic and electrochemical data of Table 1, including the appropriate electrostatic work terms43 in the calculation of the energy of the charge separated state. A strongly exergonic electron transfer pathway is clearly available for deactivation of the singlet state of the metal porphyrin unit.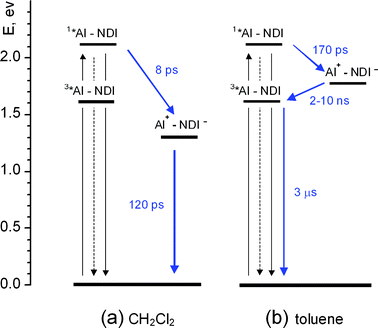 | ||
| Fig. 9 Energy level diagrams and photophysical mechanisms for the NDI–AlP dyad in dichloromethane (a) and toluene (b). Shorthand notation: AlP = Al. | ||
As a matter of fact, in dichloromethane, the naphthalenediimide and aluminium porphyrin fluorescent emissions of NDI–AlP are, respectively, completely (>99%) and strongly (ca. 90%) quenched relative to those of the model compounds. Single-photon counting experiments demonstrate that the residual emission (lifetime, ca. 5 ns) arises from impurities of unquenched aluminium porphyrin species44 whereas the fluorescence lifetime of the NDI–AlP dyad lies below the instrumental time resolution (<250 ps). The transient spectral changes observed by ultrafast spectroscopy, Fig. 10 (λexc = 590 nm, 100% AlP excitation), provide insight into the quenching mechanism. The initial spectrum is that of the excited singlet state of the aluminium porphyrin unit (see, for comparison Fig. 7a). The transient spectral changes are clearly biphasic, with a fast increase in optical density in the 580–720 nm range45 and decrease at λ > 720 nm (Fig. 10a), followed by a decay of the whole spectrum towards the baseline with isosbestic points at ΔOD = 0 (Fig. 10b). The kinetics analysis (Fig. 10c) yields time constants of 8 and 120 ps for the two processes. No long-lived transient can be observed in the nanosecond laser photolysis of NDI–AlP in dichloromethane.
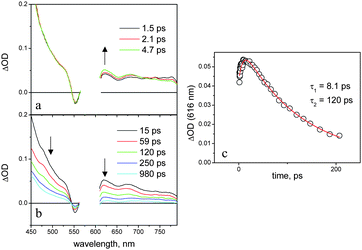 | ||
| Fig. 10 Transient absorption spectra (a, b) and kinetics (c) obtained in the femtosecond spectroscopy of NDI–AlP in dichloromethane. | ||
These spectral changes are consistent with an electron transfer quenching mechanism. The early spectral changes with time constant 8.1 ps can be assigned to charge separation, and the subsequent ones with time constant 120 ps to charge recombination. The spectral changes for the forward reaction are relatively small, as the spectrum of the AlP excited state (Fig. 7a) and that expected for a charge separated product (Fig. 8a) are not very different. However, some features of the NDI-radical anion can be recognized in the spectrum of the transient, e.g. the sharp maximum at 610 nm and the shoulder at 475 nm. The broad absorption in the 600–700 nm range, as well as the Q-band bleach at 550 nm, are signatures of the aluminium porphyrin radical cation. The photophysical behaviour of the dyad in dichloromethane is summarized in Fig. 9a.
When the solvent is changed to chloroform, the qualitative behaviour remains the same as in dichloromethane, albeit with somewhat slower kinetics: 9 and 300 ps for the first and second processes, respectively (ESI, Figure SI15†). In toluene, on the other hand, only the first process (increase in optical density in the 580–710 nm range, time constant of 170 ps) is observed in the time window (0–1 ns) of the ultrafast experiment, implying that further decay takes place in a longer ns time scale (ESI, Figure SI16†). The solvent effect on the observed kinetics is as expected for an electron transfer quenching mechanism. A decrease in solvent polarity brings about (i) a raise in energy of the charge-separated state and (ii) the decrease in reorganizational energy. For charge recombination, lying in the Marcus inverted region, both factors cooperate to slow down the rates (120 ps in dichloromethane, 300 ps in chloroform, >2 ns in toluene). For charge separation, in the less exergonic regime, the effect on rates is more complex: almost constant in dichloromethane and chloroform (8 and 9 ps), because of partial compensation between the two effects; definitely slower in toluene (170 ps), because of the prevailing effect of small driving force. The fate of the long-lived (>2 ns) charge separated state in toluene can be inferred from nanosecond laser photolysis results (ESI, Figure SI17†). The observed transient, that decays with oxygen-dependent kinetics (lifetime, 560 ns and 300 μs in aerated and deaerated solution, respectively), matches exactly the triplet state of the AlP(ba) model (Fig. 7b).
The photophysical mechanism in toluene can be schematized in the energy-level diagram of Fig. 9b (where an approximate energy of the charge separated state is obtained from the experimental redox potentials in dichloromethane, by accounting43 for the ion solvation energy differences between the two solvents). The charge-separated state is initially formed by photoinduced electron transfer in a singlet spin state. Whereas in dichloromethane charge recombination to the ground state is rapid and 100% efficient, in toluene such a process is slowed down sufficiently by inverted-region effects to allow spin inversion46 to occur in the charge-separated state. Thus, charge recombination yields the triplet state of the aluminium porphyrin unit rather than the ground state.
Photophysics of the NDI–AlMPyP–RuP triad
A simplified energy level diagram for the NDI–AlMPyP–RuP triad (high-lying excited states of the NDI unit, omitted again for clarity), as inferred from the spectroscopic and electrochemical data of Table 1, is shown in Fig. 11.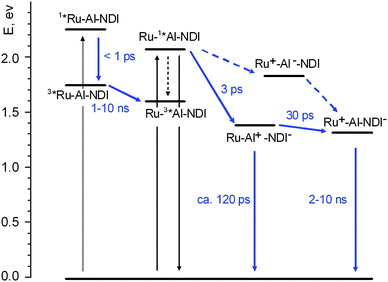 | ||
| Fig. 11 Energy level diagram and photophysical mechanisms for the NDI–AlMPyP–RuP triad in dichloromethane. Shorthand notation: AlMPyP = Al, RuP = Ru. | ||
It can be seen that a variety of quenching pathways is available following excitation of both AlP and RuP chromophoric units.
In the NDI–AlMPyP–RuP triad in dichloromethane, the naphthalenediimide and aluminium porphyrin fluorescent emissions are completely quenched relative to those of the model compounds. The transient spectral changes observed in dichloromethane by ultrafast spectroscopy (λexc = 590 nm, 95% AlP excitation) are shown in Fig. 12.
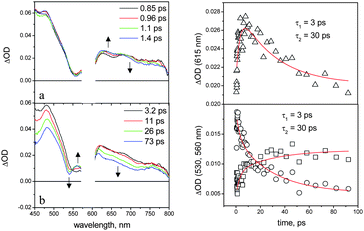 | ||
| Fig. 12 Femtosecond spectroscopy of NDI–AlMPyP–RuP in dichloromethane (590 nm excitation; for details, see text). Left: transient absorption spectra. Right: kinetics measured at 530 nm (dots), 560 nm (squares), and 615 nm (triangles). | ||
The initial spectrum is that of the excited singlet state of the aluminium porphyrin unit (see, for comparison Fig. 7a). The transient spectral changes are clearly biphasic. The relatively small, early spectral changes (time constant, 3 ps, increase in optical density in the 580–690 nm range and decrease at λ > 690 nm) resemble closely those observed for the NDI–AlP dyad (Fig. 10) and can be assigned to the same type of photoinduced electron transfer, i.e. to Ru–1*Al–NDI → Ru–Al+–NDI−primary charge separation (Fig. 11). In the subsequent time scale, however, the behaviour of the NDI–AlMPyP–RuP triad differs sharply from that of the NDI–AlP dyad. Instead of a uniform decay towards the baseline, complex spectral changes with a time constant of 30 ps are observed: aside from a moderate decrease in absorbance in the 610–800 nm range, the most evident feature is a displacement of the sharp bleach originally present at 550 nm towards 530 nm. As these wavelengths are typical of the Q-bands of the AlP and RuP units (Fig. 8a and 8b), the bleach displacement can be easily assigned to hole shift from the AlP to the RuP unit, i.e. to Ru–Al+–NDI− → Ru+–Al–NDI− secondary charge separation (Fig. 11). As a matter of fact, the final spectrum of Fig. 12 (constant in the 200–1000 ps time range, is very similar to that calculated for a charge separated state where the NDI unit is reduced and the RuP unit is oxidized (Fig. 8b) and to that experimentally observed following photoinduced charge separation in the RuP–NDI′′–RuP symmetric triad (ESI, Figure SI18†). The process assigned to charge shift (30 ps) is sufficiently fast to compete favorably with primary charge recombination, expected to take place in ca. 120 ps, as measured in the NDI–AlP dyad. That the efficiency of this step is almost quantitative is also shown by the comparable extent of the AlP and RuP features observed in the Q-band bleach shift (Fig. 12).
Considering its constancy in the time window of the ultrafast experiment, the long-range charge separated state has a lifetime longer than 2 ns. In flash photolysis experiments carried out by excitation with 590 nm, 10 ns pulses, the spectral profile of the charge separated state can be observed, although its decay could not be deconvoluted from that of the excitation pulse (ESI, Figure SI19†). Therefore, the lifetime of the long-range charge separated state lies in the few nanosecond range (2 ns < τ < 10 ns). In an attempt to observe a longer charge-separated state lifetime, some experiments were also performed for NDI–AlMPyP–RuP in toluene. In this solvent, however, the calculated energy level diagram (ESI, Figure SI20†) changes in such a way that, while primary charge separation is still appreciably exergonic, the secondary hole transfer step becomes slightly endoergonic. Indeed, the ultrafast spectral changes (ESI, Figure SI21†) do not exhibit the shift in Q-band bleach diagnostic of hole transfer, but rather the formation of the AlP triplet state.
It should be remarked that in dichloromethane an alternative electron transfer pathway is energetically available in the triad for obtaining the fully charge separated state following AlP excitation, i.e., photoinduced hole transfer from the AlP to the RuP unit, followed by electron shift from AlP to NDI (Fig. 11, dashed arrows). Along this pathway, the only process that can be associated to the 30 ps spectral shift of the bleach is photoinduced hole transfer, Ru–1*Al–NDI → Ru+–Al−–NDI. In order to follow this hypothesis, however, two rather ad hoc arguments should be introduced: (i) the early 3 ps spectral changes must simply be neglected, (ii) it must be assumed that the subsequent electron shift Ru+–Al−–NDI → Ru+–Al–NDI− is too fast to be detected. Thus, although the alternative route cannot be definitely ruled out, the most likely electron transfer pathway for deactivation of the AlP excited state is that indicated by full arrows in Fig. 11.
When the excitation wavelength is changed to 530 nm, so as to achieve substantial (ca. 80%) excitation of the RuP unit, ultrafast spectroscopy of the NDI–AlMPyP–RuP triad in dichloromethane yields the spectrum shown in Fig. 13 (delay 1.5 ps). This spectrum is clearly that of the triplet state of the RuP(py) unit (Fig. 7c). This state is formed by prompt (<1 ps) intersystem crossing and remains constant over the 1–1000 ps time scale. On the other hand, nanosecond flash photolysis (532 nm excitation) yields a clearly different spectrum (Fig. 13, delay 50 ns).
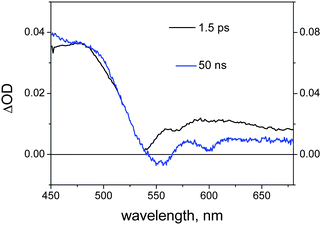 | ||
| Fig. 13 Transient absorption spectra obtained in femtosecond (black, 530 nm excitation) and nanosecond (blue, excitation at 532 nm) time-resolved spectroscopy of NDI–AlMPyP–RuP in dichloromethane. | ||
Since this spectrum matches well that of the triplet state of the AlP(ba) unit (Fig. 7b), the obvious conclusion is that efficient triplet energy transfer from the RuP to the AlP unit, 3*Ru–Al–NDI → Ru–3*Al–NDI, must occur in the “blind” time window between the two experiments (1–10 ns). The photophysical mechanism following excitation of the ruthenium porphyrin is also indicated in Fig. 11.
Conclusions
In this work, the possibility of obtaining triads for photoinduced charge separation by self-assembly of appropriate subunits has been explored using a metal-mediated strategy. An aluminium porphyrin, a ruthenium porphyrin and a naphthalenediimide unit have been used as photosensitizer, donor, and acceptor units, respectively. The key problem, i.e., how to introduce the required asymmetry into the self-assembling process, has been tackled by choosing appropriate ligand functions on the various units (carboxylate on naphthalenediimide, pyridyl on aluminium porphyrin) and exploiting hard/soft discrimination to achieve preferential coordination (carboxylate with aluminium, pyridyl with ruthenium).The self-assembly of the NDI–AlMPyP–RuP triad has been monitored by NMR spectroscopy. The three starting component, NDI, AlMPyP and RuP, in a 1:1:1 mixture self-assemble in the desired non-symmetric triad. The final photoactive three-component adduct NDI–AlMPyP–RuP is formed cleanly and quantitatively.
The actual photophysical behaviour of the NDI–AlMPyP–RuP triad has been investigated, by comparison with the NDI–AlP dyad and the individual molecular components, using time resolved spectroscopy in the fs–ps and ns time domains. The results in dichloromethane provide clear evidence for the occurrence of stepwise electron/hole transfer, leading to a charge separated state with reduced naphthalenediimide acceptor and oxidized ruthenium porphyrin donor, with a lifetime in the few nanoseconds range. Thus, the triad behaves indeed as intended by molecular design, not only with respect to self-assembling but also in terms of light-induced function.
In conclusion, this work provides an unprecedented example of a triad for photoinduced charge separation obtained by self-assembly of molecular components via coordinative bonds. The performance of the triad is clearly not exceptional in absolute terms, particularly with regard to charge separation lifetime. In fact, the system is far from being ideal in several respects (e.g., uneven energy gradients in primary and secondary electron transfer steps, conformational flexibility in the axial linkage between aluminium porphyrin and the acceptor, relatively short charge separation distance). With this type of assembling strategy, however, improvements in the performance of the triad can be simply sought by appropriate chemical modification of the individual molecular components. In principle, the key bi-functional aluminium pyridyl–porphyrin unit can be reacted with any molecular partner bearing complementary reactive sites. Thus, confining the synthetic effort to the single components, a broad choice of triad systems may become accessible. This is where self-assembling promises to be superior, in terms of ease and flexibility, to conventional covalent synthetic strategies.
Acknowledgements
Financial support from MIUR (PRIN 2006030320) (Prof. F. Scandola), EPSRC (Prof. J. K. M. Sanders) and Pembroke College (Dr G. D. Pantoş) are gratefully acknowledged. Dr E. Iengo has been supported by the EU Marie-Curie Intra-European Fellowship “EIF-Porphyrin DCLs” and by the EU Reintegration Grant “ERG-LIGHT”. We are grateful to Dr R. Argazzi for setting up the nanosecond transient absorption apparatus and to Prof. L. Hammarström, for allowing M. Orlandi to perform some flash photolysis experiments in his laboratory at Uppsala University. We thank Dr J. E. Davies of Department of Chemistry, University of Cambridge for solving the X-Ray structure of NDI′.Notes and references
- (a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS; (b) M. Hambourger, G. F. Moore, D. M. Kramer, D. Gust, A. L. Moore and T. A. Moore, Chem. Soc. Rev., 2009, 38, 25 RSC.
- (a) N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Angew. Chem., Int. Ed., 2000, 39, 1458 CrossRef CAS; (b) A. Tsuda and A. Osuka, Science, 2001, 293, 8279; (c) N. Aratani, A. D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922 CrossRef CAS.
- (a) A. Adronov, S. L. Gilat, J. M. J. Fréchet, K. Ohta, F. V. R. Neuwahl and G. R. Fleming, J. Am. Chem. Soc., 2000, 122, 1175 CrossRef CAS; (b) V. R. Frederik, F. V. R. Neuwahl, R. Righini, A. Adronov, P. R. L. Malenfant and J. M. J. Fréchet, J. Phys. Chem. B, 2001, 105, 1307 CrossRef CAS.
- (a) T. Weil, U. M. Wiesler, A. Herrmann, R. Bauer, J. Hofkens, F. C. De Schryver and K. Müllen, J. Am. Chem. Soc., 2001, 123, 8101 CrossRef CAS; (b) M. Cotlet, R. Gronheid, S. Habuchi, A. Stefan, A. Barbafina, K. Mullen, J. Hofkens and F. C. De Schryver, J. Am. Chem. Soc., 2003, 125, 13609 CrossRef CAS.
- (a) F. Puntoriero, S. Serroni, M. Galletta, A. Juris, A. Licciardello, C. Chiorboli, S. Campagna and F. Scandola, ChemPhysChem, 2005, 6, 129 CrossRef CAS; (b) F. Puntoriero, F. Nastasi, M. Cavazzini, S. Quici and S. Campagna, Coord. Chem. Rev., 2007, 251, 536 CrossRef CAS.
- J.-L. Wang, J. Yan, Z.-M. Tang, Q. Xiao, Y. Ma and J. Pei, J. Am. Chem. Soc., 2008, 130, 9952–9962 CrossRef CAS.
- (a) V. Balzani, G. Bergamini, F. Marchioni, P. Ceroni and F. Vögtle, Coord. Chem. Rev., 2007, 251, 525 CrossRef CAS; (b) C. Giansante, P. Ceroni, V. Balzani and F. Vögtle, Angew. Chem., Int. Ed., 2008, 47, 5422 CrossRef CAS.
- (a) M.-S. Choi, T. Aida, T. Yamazaki and I. Yamazaki, Angew. Chem., Int. Ed., 2001, 40, 3194 CrossRef CAS; (b) M.-S. Choi, T. Aida, T. Yamazaki and I. Yamazaki, Chem.–Eur. J., 2002, 8, 2667 CrossRef CAS.
- (a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (b) M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS; (c) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198 CrossRef CAS; (b) I. M. Bennett, H. M. Vanegas Farfano, F. Bogani, A. Primak, P. A. Lidell, L. Otero, L. Sereno, J. J. Silber, A. L. Moore, T. A. Moore and D. Gust, Nature, 2002, 420, 398 CrossRef CAS; (c) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS.
- E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147 RSC.
- (a) E. Danielson, C. M. Elliott, J. W. Merkert and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 2519 CrossRef CAS; (b) J. M. Weber, M. T. Rawls, V. J. MacKenzie, B. R. Limoges and C. M. Elliott, J. Am. Chem. Soc., 2007, 129, 313 CrossRef CAS.
- (a) D. M. Guldi, Pure Appl. Chem., 2003, 75, 1069 CrossRef CAS; (b) D. M. Guldi, H. Imahori, K. Tamaki, Y. Kashiwagi, H. Yamada, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 541 CrossRef CAS.
- (a) A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564 CrossRef CAS; (b) S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215 CrossRef CAS; (c) H. Ozawa, M. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926 CrossRef CAS; (d) P. W. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS; (e) M. Elvington, J. Brown, S. M. Arachchige and K. J. Brewer, J. Am. Chem. Soc., 2007, 129, 10644 CrossRef CAS; (f) E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378 CrossRef CAS.
- (a) A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639 CrossRef CAS; (b) A. J. Esswein, A. S. Veige and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 16641 CrossRef CAS.
- (a) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS; (b) J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954 CrossRef CAS; (c) N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS; (d) R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2008, 47, 1815 CrossRef CAS; (e) Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835 CrossRef CAS; (f) R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335 CrossRef CAS; (g) F. Jao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS.
- (a) T. A. Betley, Q. Wu, T. Van Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849 CrossRef CAS; (b) M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS; (c) M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- (a) N. D. Morris, M. Suzuki and T. E. Mallouk, J. Phys. Chem. A, 2004, 108, 9115 CrossRef CAS; (b) J. W. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS; (c) G. La Ganga, F. Nastasi, S. Campagna and F. Puntoriero, Dalton Trans., 2009, 9997 RSC.
- (a) A. Sartorel, M. Carraro, G. Scorrano, R. D. Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006 CrossRef CAS; (b) M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152 RSC; (c) Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef CAS.
- (a) C.-C. You, R. Dobrawa, C. R. Saha-Möller and F. Würthner, Top. Curr. Chem., 2005, 258, 39 CAS; (b) E. Iengo, E. Zangrando and E. Alessio, Acc. Chem. Res., 2006, 39, 841 CrossRef CAS; (c) F. Scandola, C. Chiorboli, A. Prodi, E. Iengo and E. Alessio, Coord. Chem. Rev., 2006, 250, 1471 CrossRef CAS; (d) E. Iengo, F. Scandola and E. Alessio, Struct. Bonding, 2006, 121, 105 CAS; (e) J. T. Hupp, Struct. Bonding, 2006, 121, 145 CAS; (f) H. L. Anderson, C. A. Hunter and J. K. M. Sanders, Chem. Commun., 1989, 226 RSC.
- E. Iengo, E. Zangrando, M. Bellini, E. Alessio, A. Prodi, C. Chiorboli and F. Scandola, Inorg. Chem., 2005, 44, 9752 CrossRef CAS.
- A. Prodi, M. T. Indelli, C. J. Kleverlaan, F. Scandola, E. Alessio, T. Gianferrara and L. G. Marzilli, Chem.–Eur. J., 1999, 5, 2668 CrossRef CAS.
- A. Prodi, C. Chiorboli, F. Scandola, E. Iengo and E. Alessio, ChemPhysChem, 2006, 7, 1514 CrossRef CAS.
- (a) A. Prodi, C. Chiorboli, F. Scandola, E. Iengo, E. Alessio, R. Dobrawa and F. Würthner, J. Am. Chem. Soc., 2005, 127, 1454 CrossRef CAS; (b) M. S. Rodríguez-Morgade, T. Torres, C. Atienza-Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145 CrossRef CAS.
- (a) B. G. Maiya, N. Bampos, A. A. Kumar, N. Feeder and J. K. M. Sanders, New J. Chem., 2001, 35, 797 Search PubMed; (b) J. E. Redman, N. Feeder, J. Teat and J. K. M. Sanders, Inorg. Chem., 2001, 40, 2486 CrossRef CAS; (c) J. K. M. Sanders, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic press, New York, 2000, vol. 3, pp. 347–368 Search PubMed; (d) H.-J. Kim, N. Bampos and J. K. M. Sanders, J. Am. Chem. Soc., 1999, 121, 8120 CrossRef CAS.
- (a) G. J. E. Davidson, L. H. Tong, P. R. Raithby and J. K. M. Sanders, Chem. Commun., 2006, 3087 RSC; (b) G. A. Metselaar, J. K. M. Sanders and J. de Mendoza, Dalton Trans., 2008, 588 RSC; (c) G. J. E. Davidson, L. A. Lane, P. R. Raithby, J. E. Warren, C. V. Robinson and J. K. M. Sanders, Inorg. Chem., 2008, 47, 8721 CrossRef CAS; (d) M. S. Miller, H. L. Filiatrault, G. J. E. Davidson, M. Luo and T. Breen Carmichael, J. Am. Chem. Soc., 2010, 132, 765 CrossRef CAS.
- A number of triads assembled by metal coordination have been described. They are, however, either symmetric systems (i.e., pseudo-diads)29 or triads in which two out of three molecular components are covalently linked.30 Non-symmetric triads assembled by means of other non-covalent strategies have been reported, see for example: (a) R. Ballardini, V. Balzani, M. Clemente-León, A. Credi, M. T. Gandolfi, E. Ishow, J. Perkins, J. F. Stoddart, H.-R. Tseng and S. Wenger, J. Am. Chem. Soc., 2002, 124, 12786 CrossRef; (b) B. Ferrer, G. Rogez, A. Credi, R. Ballardini, M. T. Gandolfi, V. Balzani, Y. Liu, H.-R. Tseng and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18411 CrossRef CAS.
- e.g., (a) R. Dobrawa and F. Würthner, Chem. Commun., 2002, 1878 RSC; (b) A. Harriman, J. P. Rostron, M. Cesario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110, 7994 CrossRef CAS; (c) Y. Terazono, G. Kodis, P. A. Liddell, V. Garg, M. Gervaldo, T. A. Moore, A. L. Moore and D. Gust, Photochem. Photobiol., 2007, 83, 464 CAS.
- (a) F. D'Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898 CrossRef CAS; (b) F. D'Souza, M. E. El-Khouly, S. Gadde, M. E. Zandler, A. L. McCarty, Y. Araki and O. Ito, Tetrahedron, 2006, 62, 1967 CrossRef CAS; (c) P. K. Poddutoori, A. S. D. Sandanayaka, T. Hasobe, O. Ito and A. van der Est, J. Phys. Chem. B, 2010, 114, 14348 CrossRef CAS.
- J. K. M. Sanders, N. Bampos, Z. Clyde-Watson, S. L. Darling, J. C. Hawley, H.-J. Kim, C. C. Mak and S. J. Webb in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic press, New York, 2000, vol. 3, pp. 1–48 Search PubMed.
- RuP – NDI ′′– RuP is clearly a triad from a structural viewpoint. From the functional viewpoint, however, symmetric triads can be effectively considered as pseudo-dyads.
- Free NDI is barely soluble in chlorinated solvents.
- An association constant of ca. 3 × 10−3 M−1 was estimated for 4-tert-butylpyridine binding to an aluminium tetraphenyl porphyrin.27a.
- (a) C. A. Hunter and L. D. Sarson, Angew. Chem., Int. Ed. Engl., 1994, 33, 2313 CrossRef; (b) X. Chi, A. J. Guerin, R. A. Haycock, C. A. Hunter and L. D. Sarson, J. Chem. Soc., Chem. Commun., 1995, 2563 RSC; (c) X. Chi, A. J. Guerin, R. A. Haycock, C. A. Hunter and L. D. Sarson, J. Chem. Soc., Chem. Commun., 1995, 2567 RSC.
- A very similar behaviour is shown by the model compound AlMPyP(ba), in which the –OH has been replaced with benzoic acid, in chloroform and pyridine solutions (see Scheme SI1 and Figure SI8, ESI†). The self-assembly of this species is also observed in the absorption spectrum, as a broadening of the main Q-band at 550 nm and a red shift (ca. 10 nm) of the 590 nm band (ESI, Figure SI13†). The effect is concentration dependent and practically disappears for <5 × 10−5 M solutions.
- M. T. Indelli, C. Chiorboli, M. Ghirotti, M. Orlandi, F. Scandola and H. J. Kim, J. Phys. Chem. B, 2010, 114, 14273 CrossRef CAS.
- For AlMPyP(ba), no self-association36 takes place at the concentrations used in the spectroscopic investigations.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS.
- This indicates that the self-association36 has little effect on the reduction potential of this species.
- S. Alp, U. Erten, C. Karapire, B. Köz, A. O. Doroshenko and S. Içli, J. Photochem. Photobiol., A, 2000, 135, 103 CrossRef CAS.
- Relative intensities of the component radical ion spectra from spectroelectrochemical experiments on equal concentration solutions of the AlP(ba), NDI′ and RuP(py) models.
- (a) D. Rehm and A. Weller, Ber. Bunsen- Ges. Phys. Chem., 1969, 73, 834 CAS; (b) A. Weller, Z. Phys. Chem., 1982, 133, 93 CAS . For the NDI–AlP dyad, a centre-to-centre distance of 8.5 Å, as obtained from the DFT-optimized structure, has been used. For the NDI–AlMPyP–RuP triad, in which the conformational freedom allows for a range of centre-to-centre distances, an average value of 12 Å has been assumed. Effective radii of 4 and 5 Å were assumed, respectively, for the NDI and porphyrin molecular fragments.
- The unquenched emission indicates the presence of aluminium porphyrin lacking the NDI coordinated group. In principle, this could arise from (i) an equilibrium between coordinated and uncoordinated species or (ii) an excess of free aluminium porphyrin from the synthesis. The fact that a proportion of unquenched emission is constant as a function of concentration (in the 5 × 10−6–6 × 10−5 M range) rules out hypothesis (i). Furthermore, hypothesis (ii) is consistent with a quantitative analysis of the absorption spectra of Fig. 6, which reveals a small (ca. 10% excess) of porphyrin over NDI spectral features.
- The 570–610 nm region, hidden in Fig. 10 by the excitation pulse, can be monitored by using 550 nm as excitation wavelength (see Figure SI14, ESI†).
- T. van der Boom, R. T. Hayes, Y. Zhao, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 9582 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental Section; 1H NMR of NDI, NDI′, NDI′′–RuP–NDI′′, AlMPyP(ba); H–H COSY of NDI–AlP, RuP–NDI′′–RuP, AlMPyP, AlMPyP(ba), AlMPyP–Ru, NDI–AlMPyP–RuP; absorption spectrum of AlMPyP(ba); transient absorption spectra of NDI–AlP, RuP–NDI′′–RuP, NDI–AlMPyP–RuP; X-ray structure of NDI′ with crystal data. CCDC reference number804310. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00520g |
| ‡ Up to now, attempts to obtain mass spectra (Electrospray or MALDI) of the adducts were not successful. |
| § The presence of tert-butyl groups on the phenyls on both the metallo-porphyrins is essential to give a wider range of solubility to the assembled systems. Moreover, the different positions of these alkyl substituents (in meta and in para for the aluminium porphyrins and for the ruthenium porphyrin, respectively) allows for a better resolution and easier attribution of the resonances in the aromatic region of the 1H NMR. |
| This journal is © The Royal Society of Chemistry 2011 |