The bigger, the better: Ring-size effects of macrocyclic oligomeric Co(III)-salen catalysts†
Yu
Liu
a,
Jonathan
Rawlston
b,
Andrew T.
Swann
b,
Tait
Takatani
c,
C. David
Sherrill
c,
Peter J.
Ludovice
*b and
Marcus
Weck
*a
aMolecular Design Institute and Department of Chemistry, New York University, New York, NY 10003-6688, USA. E-mail: marcus.weck@nyu.edu
bSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, 30332, USA. E-mail: pete.ludovice@chbe.gatech.edu
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 17th December 2010
Abstract
Macrocyclic oligomeric Co(III)-salen complexes derived from cyclooctene salen monomers are among the most active catalysts for asymmetric epoxide ring-opening reactions. Due to the uncontrollable feature of the ring-expanding olefin metathesis step during catalyst synthesis, the macrocyclic oligomeric Co(III)-salen complexes are produced as mixtures of oligomers with different ring sizes. We rationalize that the ring size of the Co(III)-salen oligomers might have a significant effect on the catalytic efficiency and selectivity and report here a purification protocol to isolate macrocyclic dimers, trimers, tetramers and oligomeric mixtures with larger size rings. Hydrolytic kinetic resolution (HKR) tests using allyl glycidyl ether as substrate show that the dimer is inactive at a catalyst loading of 0.01 mol%. Increasing ring size shows a remarkable effect on reaction rates with the largest ring-size species exhibiting superior selectivities and activities. NMR studies reveal that the dimeric catalyst is strained which is not observed for the larger ring-size catalysts. Computational modeling studies indicate that the dimer is lacking the flexibility to allow adjacent Co(III)-salen groups to form a bimetallic complex. Further catalytic tests of larger ring-size Co(III)-salen complexes (tetramer to hexamer mixture) by investigating the HKR of various racemic terminal epoxides and the asymmetric epoxide ring-opening with different nucleophiles demonstrate the superior catalytic activity of large ring-size macrocyclic catalysts. Furthermore, this study demonstrates again the structural (or configurational) sensitivity of Co(III)-salen catalyst towards the selectivity and efficiency of cooperative bimetallic reactions.
Introductions
Enzyme specificity and reactivity are governed by many factors including the size of the substrate binding pocket which is an obvious target for mutagenesis aimed at improving or modifying enzyme properties.1 To mimic enzyme functions, many natural or synthetic macrocyclic oligomers have been developed including cyclodextrins, crown ethers, cyclophanes, calixarenes and cyclic porphyrin.2 These oligomers and their derivatives exhibit impressive catalytic activity in many organic reactions.3–5 Much like enzymes, the ring size of these macrocyclic oligomer based catalysts has a great impact on recognition and the reaction rates6 and can influence the stereoselectivity of the products.7 In these cases, while the cyclic structure brings two or more functional moieties in close proximity, only an optimized ring size allows for all functional moieties to reach the ideal range of distance, orientation and position to interact with the reactants selectively. In this contribution, we report that cyclic Co(III)-salen complexes follow these guidelines. Through optimization of ring-cycle size, superior catalysts can be obtained for the hydrolytic kinetic resolution (HKR) of epoxides.The asymmetric ring-opening of epoxides catalyzed by chiral metal-salen complexes has emerged as a powerful method to resolve racemic epoxides and/or prepare chiral diols or alcohols.8 With water as the nucleophile, the HKR of terminal epoxides catalyzed by Co(III)-salen complexes such as 1 is featured with exceptional enantioselectivity, quantitative yields and mild reaction conditions.9,10 Detailed kinetic studies have revealed a coordinated bimetallic mechanism of the HKR, in which one Co(III)-salen moiety presumably activates the epoxides as a Lewis acid while the other Co(III)-salen binds to hydroxide.11 To enhance the cooperativity of the catalytic centers and also to develop more practical and recyclable catalytic systems, we and others have developed supported versions of 1 (Fig. 1) using a variety of supports including polymers,12–17dendrimers,18 oligomers,19,20silica,12 resins,21zeolites,22 and nanoparticles.23 To date, the two most active Co(III)-salen systems for the HKR are based on macrocyclic oligomeric structures.24Co(III)-salen complex 2 reported by Jacobsen and co-workers has the Co(III)-salen moieties embedded in a macrocyclic lactone framework.19,25–27 We reported the ring-expanding olefin metathesis of cyclooctene salen monomers to generate Co(III)-salen complex 3, which has the pendent Co(III)-salen units tethered on a macrocyclic ring.20 Both cyclic salen systems are similar in activities and selectivities through enforcement of the cooperative bimetallic mechanism. However, due to the uncontrollable features of both cyclization reactions, these support systems are produced as mixtures of macrocyclic oligomers of different ring sizes. For our Co(III)-salen catalyst 3, MALDI-TOF mass spectra revealed a mixture of compositions ranging from cyclic dimers to cyclic decamers with decreasing intensity from low molecular species to high molecular species.
 | ||
| Fig. 1 Monomeric and macrocyclic Co(III)-salen complexes. | ||
It has been suggested in the literature that the activity and selectivity of a cyclic catalytic system can be dependent on the ring size of the cycle. For example, Hine and Flachska have reported that the catalytic activities during the dedeuteration of aldehyde-2-d using poly(ethylenimine)s (PEI) depend highly on the molecular weight of the PEI and that higher molecular weight samples are more active.28 The authors suggest that this phenomenon was due to the larger number of amino groups available for intramolecular catalysis in the high molecular weight PEI.
In addition, other factors, such as the configuration and/or arrangement of catalytic groups, can have a substantial impact on the catalytic efficiency and even the reaction pathway, especially for cooperative catalytic systems. For instance, Katz and co-workers have compared acid–base bifunctional effects in homogeneous and heterogeneous catalysts for the Henry reaction. Cooperativity between catalytic sites was not observed for the homogeneous catalyst. However, it was observed for the heterogeneous system, although the acid and amine groups are in similar stoichiometric proportions and close proximity for both systems.29,30 Owing to its bimetallic coordinated mechanism, Co(III)-salen catalyzed asymmetric ring-opening reactions are also sensitive to orientation, distance and flexibility of the salen moieties.31,32 It was reported that the ring size of the macrocyclic Co(III)-salen complex 2 has a strong influence on the catalytic activity in the order of trimer > dimer > tetramer.25 Therefore, we rationalize that the ring size of our Co(III)-salen oligomers 3 will also have a significant effect on the catalytic efficiency and selectivity. Superior catalytic activities and selectivities are expected to be achieved through isolating the most active and selective Co(III)-salen cycle.
In this contribution, we report a method to separate our cyclooctene-based macrocyclic salen ligands based on ring size. The catalytic activities and selectivities of the isolated cyclic oligomers were, after metalation, compared for the HKR of allyl glycidyl ether. Our results demonstrate that different ring-size oligomers exhibit dramatically different catalytic behaviors. At the same catalyst loading (0.01 mol%), the dimeric species almost did not show any catalytic activity. In contrast to 2 where trimers are the most reactive species while larger ring-cycles (tetramers) are significantly less active, a remarkable enhancement of the catalytic activity of our isolated cycles goes hand in hand with increasing ring size resulting in superior catalytic performance by oligomers with larger ring-sizes. NMR spectroscopy and computational methods were employed to rationalize these different catalytic behaviors from a structural point of view. The large ring-size catalysts (tetramer to hexamer mixture) were further tested for the HKR of a variety of terminal epoxides and the asymmetric epoxide ring-opening with other nucleophiles than water. Compared to our original oligomeric mixture, the isolated larger cyclic Co(III)-salen oligomers demonstrate an exceptional increase in reactivity, enantioselectivity, and reaction scope making them the most active and selective Co(III)-salen catalysts to date.
Results and discussion
Preparation of macrocyclic Co(III)-salen complexes with different ring sizes
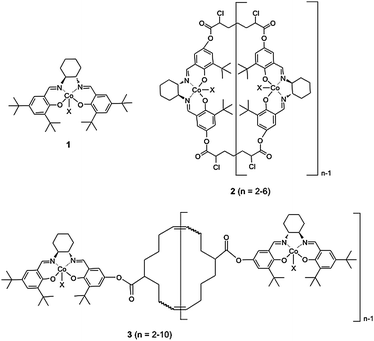
The macrocyclic salen ligands were synthesized as described in our previous report and were obtained as a mixture of different ring-size oligomers.20 MALDI-TOF mass spectrometry (MALDI-TOF MS) of the oligomeric mixture showed predominantly dimers and trimers. With increasing ring size, the intensities of the mass signals gradually decreased until nonamers and decamers are barely detectable (ESI†). The separation of the macrocyclic oligomers started with a silica chromatography purification step using a hexane–ethyl acetate mixture containing 0.2% triethyl amine as the eluent. Individual fractions were analyzed by MALDI-TOF MS. The dimer eluted first followed by medium ring size oligomers and finally larger macrocyclic oligomers. The silica column chromatography step yielded pure dimer but all other ring-size oligomers were still obtained as mixtures. All fractions but the pure dimer were further subjected to size-exclusive chromatography (SEC) purifications. Because of its organic solvents compatibility, the Toyopearl HW40 resin was employed for the SEC separation step. Several solvents or solvent combinations were examined including chloroform, THF, DMF, methanol and methanol–chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Chloroform and THF did not afford good separation of the different sized salen oligomer ligands while the oligomers were not fully soluble in methanol. Both DMF and the methanol–chloroform mixture showed good separation based on ring size and the methanol–chloroform mixture was chosen as the final eluent. The SEC fractions were analyzed by MALDI-TOF MS. During the SEC purification step, larger oligomers were eluted out first. After one round of purification, pure trimer could be obtained. Further rounds of SEC purification afforded tetramer contaminated with a trace amount of trimer and pentamer (Fig. 2A). For higher mass oligomers, SEC yielded only larger oligomer mixtures with complete removal of the lower mass oligomers. In the end, the following oligomer fractions were obtained: dimers, trimers, tetramers, tetramers to hexamers and pentamers to decamers (Fig. 2A). These fractions were also characterized by gel-permeation chromatography (GPC) (ESI†). Although the GPC was calibrated versus linear poly(styrene) rather than structurally analogous macrocyclic oligomers, trimers, tetramers, tetramers to hexamers and pentamers to decamers all showed number-average molecular weights (Mn) close to the theoretical data excepted for the dimers which shows a higher Mn than the theoretical mass. Since the separated species are either a single type oligomer or oligomers with a narrow ring size distribution, small polydispersity indices (PDIs) are observed (1.06 to 1.12).
:1). Chloroform and THF did not afford good separation of the different sized salen oligomer ligands while the oligomers were not fully soluble in methanol. Both DMF and the methanol–chloroform mixture showed good separation based on ring size and the methanol–chloroform mixture was chosen as the final eluent. The SEC fractions were analyzed by MALDI-TOF MS. During the SEC purification step, larger oligomers were eluted out first. After one round of purification, pure trimer could be obtained. Further rounds of SEC purification afforded tetramer contaminated with a trace amount of trimer and pentamer (Fig. 2A). For higher mass oligomers, SEC yielded only larger oligomer mixtures with complete removal of the lower mass oligomers. In the end, the following oligomer fractions were obtained: dimers, trimers, tetramers, tetramers to hexamers and pentamers to decamers (Fig. 2A). These fractions were also characterized by gel-permeation chromatography (GPC) (ESI†). Although the GPC was calibrated versus linear poly(styrene) rather than structurally analogous macrocyclic oligomers, trimers, tetramers, tetramers to hexamers and pentamers to decamers all showed number-average molecular weights (Mn) close to the theoretical data excepted for the dimers which shows a higher Mn than the theoretical mass. Since the separated species are either a single type oligomer or oligomers with a narrow ring size distribution, small polydispersity indices (PDIs) are observed (1.06 to 1.12).
 | ||
| Fig. 2 MALDI-TOF mass spectra of the separated macrocyclic oligomeric salen ligands (A) and corresponding Co(II)-salen complexes (B). | ||
Under an inert atmosphere, the macrocyclic ligands, on treatment with Co(OAc)2·4H2O in methanol, were converted to the corresponding Co(II)-salen complexes. The metalated products were generated in 92%–97% yields as a brick red powder. Fig. 2B shows the MALDI-TOF mass spectrum of the Co(II)-salen complexes. The elemental analyses of all Co(II)-salen complexes gave cobalt loadings from 8.03% to 7.38% (w/w), indicating metalation yields of 95.4% to 87.6%. The Co(II)-salen complexes were converted to the catalytically active Co(III)-salen complexes by aerobic oxidation in the presence of acetic acid. After azeotropic drying with toluene and vacuum drying to remove any acetic acid residue, all the Co(III)-salen oligomeric catalysts were obtained as a dark brown solid.
HKR using racemic allyl glycidyl ether
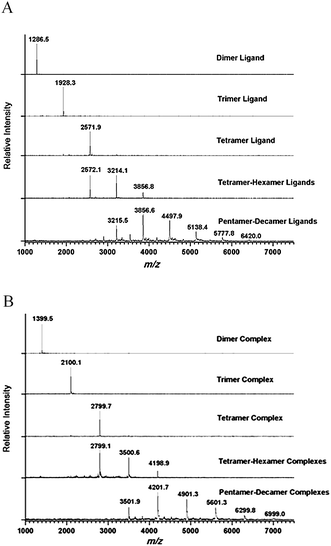
The HKR of allyl glycidyl ether was chosen as the model reaction to evaluate the catalytic activity of the macrocyclic Co(III)-salen complexes with different ring sizes. The resolutions were carried out at ambient temperatures with 0.6 equiv of water and a 0.01 mol% catalyst loadings based on cobalt. Reaction progress was monitored by chiral GC analysis. The results are shown in Fig. 3. The catalytic activity and selectivity of the original oligomeric mixture is shown as standard.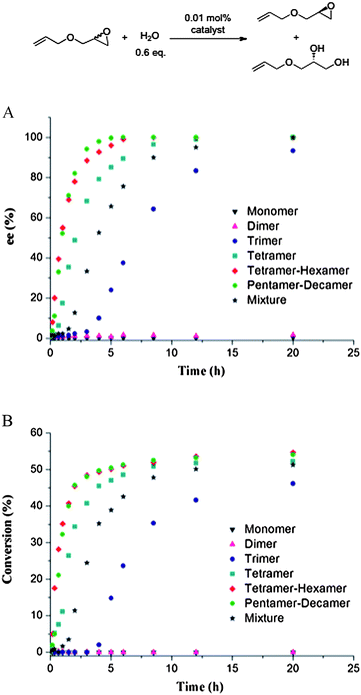 | ||
| Fig. 3 HKR of allyl glycidyl ether catalyzed by macrocyclic Co(III)-salen complexes 3 of different ring sizes (n = 1, 2, 3, 4, 4∼6, 5∼10 and mixture). Kinetic plots of (A) enantiomeric excess and (B) conversion. | ||
As expected, the Co(III)-salen cyclooctene monomer did not afford any resolved product indicating the lack of cooperation between salen species at such a low catalyst concentration. The dimeric Co(III)-salen complex showed almost no catalytic effect either. After 20 h, less than 2% enantiomeric excess (ee) was observed with no detectable conversion. The trimer system has a very long incubation period. Two hours after the addition of water, the initial rate is still close to zero. Eventually, 94% ee and 46% conversion were reached in 20 h making the trimers significantly less active and selective than our original oligomeric mixture. With increasing ring size, the reaction rate further increases. The tetramer species is already more active than the original mixture finishing the reaction in 12 h (>99% ee, 52% conversion). The larger ring-size Co(III)-salen oligomers are superior catalysts. The tetramer-hexamer mixture afforded >99% ee and 51% conversion of the epoxide in 6 h. Although the pentamer-decamer mixture has a lower initial rate than tetramer-hexamer mixture (0.0042 mmol s−1vs. 0.0053 mmol s−1), the pentamer-decamer mixture exhibited the best catalytic efficiency giving above 99% ee and 50.4% conversion in 5 h. In comparison, the original Co(III)-salen complex mixture provided the resolved target epoxide in 95% ee and 50% conversions after 12 h reaction. The larger size catalytic system (pentamer-decamer) shortened the reaction time by more than half in comparison to the original mixture.
NMR studies of the macrocyclic salen oligomers with different ring sizes
Next, we carried out a series of experiments to understand this significant increase in reactivity for the larger ring sizes. We obtained 1H NMR spectra of all macrocyclic salen ligands and the salen monomer (Fig. 4). While the monomer has sharp, well-defined and split NMR signals, the corresponding signals broaden in the spectra of the macrocyclic oligomers. Interestingly, the 1H-NMR spectrum of the dimer shows significant differences to all other cyclic oligomers. First, some NMR signals in the dimer spectra are split in comparison to the larger size oligomers including the phenol O–H signal at 13.9 ppm, the two N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H at 8.25 ppm and 8.34 ppm, the aromatic proton at 6.75 ppm and the tertiary C–H from 2.4 ppm to 2.9 ppm. For the phenol O–H at 13.65 ppm, the NC–H at 8.25 ppm and the aromatic proton at 6.75 ppm the splitting is accompanied with the appearance of a tiny side peak. In the latter two protons, the side peaks are shifted by more than 0.1 ppm upfield relative to the main signal. When carefully integrating the 1H NMR spectrum of the dimer, all split peaks (the side peaks were integrated together with the main signals) can be individually integrate as one proton (see ESI†). The second major difference is the alkenyl protons at 5.4 ppm. For all higher mass oligomers, the corresponding signals have exactly the same chemical shift and overlap perfectly. However, the alkenyl peak of the dimer is shifted about 0.1 ppm downfield in comparison to the higher mass oligomers.
C–H at 8.25 ppm and 8.34 ppm, the aromatic proton at 6.75 ppm and the tertiary C–H from 2.4 ppm to 2.9 ppm. For the phenol O–H at 13.65 ppm, the NC–H at 8.25 ppm and the aromatic proton at 6.75 ppm the splitting is accompanied with the appearance of a tiny side peak. In the latter two protons, the side peaks are shifted by more than 0.1 ppm upfield relative to the main signal. When carefully integrating the 1H NMR spectrum of the dimer, all split peaks (the side peaks were integrated together with the main signals) can be individually integrate as one proton (see ESI†). The second major difference is the alkenyl protons at 5.4 ppm. For all higher mass oligomers, the corresponding signals have exactly the same chemical shift and overlap perfectly. However, the alkenyl peak of the dimer is shifted about 0.1 ppm downfield in comparison to the higher mass oligomers.
 | ||
| Fig. 4 1H NMR (400 MHZ, CDCl3, room temperature) comparison of the cyclooctene salen monomer, the cyclic salen dimer, the cyclic salen trimer, the cyclic salen tetramer and finally the cyclic salen pentamer-decamer mixture. Split peaks are marked with an asterisk. | ||
To further investigate the different splitting and chemical shifts in the 1H NMR spectra of the dimer, we carried out 1H-13C HSQC measurements. As can be seen from Fig. 5A, the two NC–H signals stem from two carbons, respectively, and the split aromatic proton at 6.75 ppm stems from a single carbon. Together with the integration results, these observations verified that the split signals and side peaks are from the dimer molecule and that sample contaminations can be excluded.
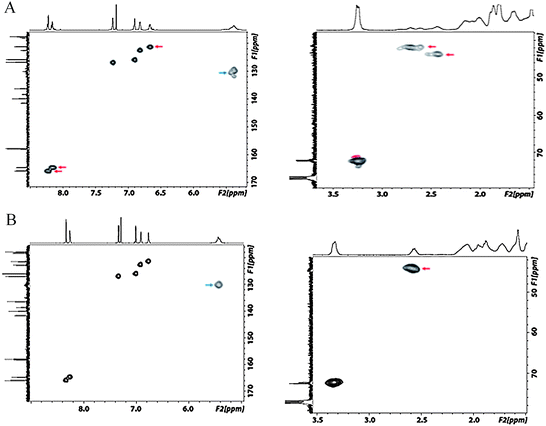 | ||
| Fig. 5 Partial 1H-13C HSQC spectra (400 MHZ, CDCl3, room temperature) of the macrocyclic dimer salen ligand (A) and the tetramer-hexamer salen ligand mixture (B). The signals discussed are indicated by arrows. | ||
Some differences for the tertiary carbon f were also observed (see Fig. 4 and 5). For the dimer ligand, both, the proton signal (2.4 ppm to 2.9 ppm) and the corresponding carbon signal (41.5 ppm to 44.0 ppm), are highly split and result in multiple diagonal peaks in the HSQC spectrum. In contrast, for the tetramer to hexamer mixture, only a single diagonal peak of this tertiary carbon is detected (Fig. 5B). Finally, the alkenyl carbon e/e′ at 130 ppm shows also a strong ring size dependence. In the 1H-13C HSQC spectrum of the dimer this carbon signal is severely split in comparison to a broad but single diagonal signal of the tetramer to hexamer mixture.
The 1H, 13C, and 1H-13C HSQC NMR experiments indicate that the dimer salen ligand has a different chemical environment than all other higher mass oligomers. During the ring-expanding olefin metathesis step, the ring-strain of the cyclooctene is released by forming a larger macrocyclic ring, which leads to an upfield shift of the alkenyl proton peaks. The alkenyl peak of the dimer does not overlap with those of the larger mass oligomers suggesting that some weak strain might exist in the dimer. Theoretically, the dimer has three different isomers based on the configurations of the two alkenyl bonds in the macrocyclic backbone: cis-cis, cis-trans and trans-trans. All three isomers might be in different chemical environments, resulting in slightly different chemical shifts and/or the split signals.33 The tertiary carbon f is a chiral carbon, i.e. diastereomers will be generated during the synthesis of the macrocyclic oligomers. The number of diastereomers depends on the number of tertiary carbons in different ring-size oligomers. In most cases, one expects the chemical differences of the diastereomers to be negligible due to the high degree of similarity of the two alkyl substituents of the chiral center. Therefore, this tertiary C–H only shows a broad signal at 2.55 ppm for the trimer and all larger ring-size oligomers. However, for the dimer, because of the strained ring structure and the large salen pendants, this diastereotopic difference is probably amplified leading to highly split signals observed in both the 1H NMR and the 13C NMR spectra of this tertiary carbon.
In order to obtain further information about the structural aspect of the dimer ligand, Variable Temperature NMR (VT-NMR) experiments (20 °C to 90 °C) were performed in tetrachloroethene-d2 (Fig. 6). The coalescence of the well-defined split proton signals into broad signals at higher temperature was observed for the two NC–Hprotons and the aromatic proton at 6.75 ppm. Because of the easy exchange of the hydroxyl proton at high temperature, the two phenol O–H peaks were fully merged into one broad signal at 80 °C. The two aromatic signals at 6.9 ppm and 7.3 ppm shifted upfield with rising temperature, which leads the 6.9 ppm peak to merge with another aromatic peak at 7.0 ppm at 90 °C. No coalescence or shift of the tertiary C–H from 2.5 ppm to 2.9 ppm was observed, which probably indicates the configuration of this tertiary carbon is stable under the thermal conditions of the VT-NMR experiment. These experiments provide some hints about the structure of the dimer. Our hypothesis is that at ambient temperature, the interconversion of the three isomers is prohibited due to an energy barrier, i.e. the proton signals of individual isomers can be detected. With increasing temperature, the thermal transformation of the configurations occurs and results in isomer exchanges.34 Eventually, the coalescence of split peaks could be observed at high temperature.
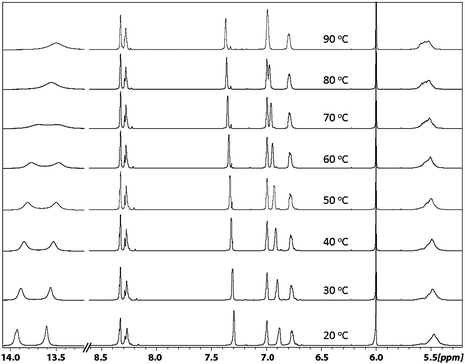 | ||
| Fig. 6 Variable-temperature NMR spectra (400 MHz, tetrachloroethene-d2) of the macrocyclic dimer salen ligand. | ||
The combined NMR studies allow for a couple of basic predictions. With regard to catalytic activity of the corresponding Co(III)-salen complexes in the hydrolytic kinetic resolution of epoxides, we expect that the Co(III)-salen dimer, as a result of their relatively strained small ring structure, might have more difficulty in reaching the ideal conformation for the bimetallic pathway reaction without severely distorting the ring structure. The result would be a less active catalyst. When the ring size increases to a trimer, more flexibility was obtained leading to a remarkable enhancement of catalytic efficiency. However, some rigidity might still exist in the trimer structure. Longer times and/or higher energy might be needed to reach the ideal conformation for bimetallic processes. For larger ring-sizes, we expect that the macrocycles are highly flexible endowing the salen moieties with superior catalytic activity. Another factor, which cannot be excluded, is the slow counterion exchange of trimer to generate the active Co(III)-hydroxide which has been proposed by Blackmond and Jacobsen.11
Molecular modeling of the macrocyclic Co(III)-salen complexes
We next investigated the structural composition of the different oligomeric cycles via computer simulation to gain further understanding of the origin of their different catalytic behaviors. The fully metalated species, i.e.Co(III)-salen macrocyclic oligomers with acetate (OAc) as counter-ion, was modeled for the dimer and trimer in various configurational isomers using molecular dynamics (MD). This MD trajectory was then sampled to determine how often the Co-salen groups were within a reasonable proximity of each other to approximate the formation of the bimetallic complex. The relative probability of this complex formation was assumed to approximate the relative probability of the bimetallic reaction.The MMFF9435 force field was applied to simulate the macrocyclic octene backbone. It was also used to describe most of the force field for the outer atoms of the Co-salen ligand. However, the MMFF force field does not contain parameters for cobalt. Therefore the force field parameters for Co and its adjacent atoms in the ligand were taken from the extensible systematic force field (ESFF).36 What constitutes the adjacent atoms is described in the ESI.† The ESFF force field was parameterized for organometallic molecules and had been used previously to model the manganese center of the Mn-salen complex.37 Because the bonded force field equations for MMFF and ESFF differ slightly in their form, the bonded contribution of ESFF was fit to the form of the MMFF equations. The same simplifications used in ESFF were also used in the resulting fitted MMFF force field. These simplifications result in all the torsional angle parameters involving the Co atoms being set to zero. Additionally, the ESFF force field does not have defined cross terms or out-of-plane energies for metal atoms, so these can also be set to zero. This means that the only parameterization required is for bond stretch, angle bend, van der Waals, and electrostatic interactions. Of these, the ESFF force field will be used to parameterize the MMFF form of all of these interaction energies except for the electrostatic interactions which were derived from a DFT calculation below.
While the force field bonded and van der Waals parameters were used for the center of the Co-salen complex as described above, the equilibrium bond lengths and bond angles were taken from a structure of the complex determined by X-ray diffraction. Although a number of Co(III)-salen X-ray diffraction structures have been published in recent years,38–40 there are no available X-ray diffraction data on a structure using the acetate counter-ion. The aforementioned bond length and bond angle parameters were taken from the structure reported by Cohen and coworkers on Co(III)-salen which utilizes a chlorine counter-ion.40 The remaining parameters for the acetate counter ion, including the static partial atomic charges for all the atoms in the ligand, were determined from the ab initio quantum calculation of the Co-salen ligand described below.
The ab initio calculation that was used as the basis for the force field parameterization was performed on a reduced Co-salen structure that is essentially the structure pictured in Fig. 7 with all t-butyl groups removed. These groups were removed as a computational expedient. Density functional theory (DFT), executed with the Jaguar suite of programs (Jaguar 5.5, Schrödinger, LLC, Portland OR, USA), was used to compute the optimized singlet state structure of the Co(III) metal-salen model with the BP86 functional and the LACVP* basis set (LAN2DZ for Cobalt and 6-31G* for all other atoms). A frequency computation at the converged geometry was performed to ensure the structure corresponded to a potential energy minimum. Atomic charges were computed by fitting to the DFT electrostatic potential. The coordinates and partial charges resulting from this computation can be found in Appendix A of the Supplementary Information† and these partial charges were used for all atoms in the Co(III)-salen complex to provide consistency. The fact that t-butyl groups were missing from this DFT calculation does not affect the result as the atoms in these alkane groups are assigned partial charges of zero. The acetate group bonded force field parameters, as well as their equilibrium bond lengths and angles were taken from the energy minimized geometry of this DFT calculation, because no X-ray diffraction structure was available with the acetate ion. The MMFF force field equations were fit to the ESFF energies for the atoms adjacent to the Co metal center to obtain parameters for the MMFF force field equations that mimic the ESFF force field. Similarly, the MMFF force field equations were fit to the geometry and Hessian matrix of the DFT equations. The resulting parameters from this extended version of the MMFF force field are listed in the ESI.†
 | ||
| Fig. 7 Co(III)salen complex with acetate counter-ion. | ||
The hybrid force field described above was used in MD calculations to examine the likelihood of the Co(III)-salen ligands forming a bimetallic complex. This likelihood of complex formation was then assumed to be proportional to the reactivity of the particular isomers. MD was used to sample the conformation space of the oligomers at 300 K using the Nosé-Hoover algorithm41 with a thermal coupling parameter of 0.5 kcal ps Å−1. The time step was 0.001 ps, and the hydrogen bond lengths were constrained using the LINCS constraint algorithm.23 The simulation was sampled every 0.1 ps. The simulations were run for 2 ns, and structures were sampled ever 0.5 ps for analysis of the formation of complexes. A minimum separation distance of 10 Å between cobalt centers was required for a bimetallic complex to be counted,42 as well as that the Co-salen planes deviated no more than 30° from parallel and the head-to-tail vectors of the planes deviated no more than 45° from anti-parallel. An implicit solvent model was used to approximate solvent interactions on the system. The solvent model used was the generalized Born implicit solvent model,43 and the dielectric constant was set to that of ethylene oxide, which is 14.44Ethylene oxide was used because it is the smallest terminal epoxide that might be used with these catalysts.
Using the criterion above for the formation of bimetallic complex, the dimer, regardless of the cis or trans distribution, never formed a complex. Fig. 8 shows the cumulative number of complexes formed during the MD simulation for various configurational isomers in the trimer. Initial structures were formed by adding a closing harmonic constraint to the ends of a liner molecule built in the Molecular Operating Environment (MOE) from Chemical Computing Group (http://www.chemcomp.com). MOE was also used for the MD simulation with the force field discussed above. An energy minimization was performed to an energy gradient tolerance of 0.1 kcal/mole Å with an applied harmonic constraint. A subsequent energy minimization was carried out after replacing the constraint with an actual C–C bond. Fig. 8 shows the typical results that after 1 ns a linear region is observed. Therefore the first ns of the simulation was discarded and only the final ns of the simulation was analyzed to determine the likelihood of complex formation as seen in Fig. 9. Each point in both Fig. 8 and 9 correspond to an average of 40 separate simulations. Each simulation used the same initial conformation, but used a different random set of velocity vectors. The error bars in Fig. 9 are the 90% confidence intervals about those points. Only a few error bars were added, but these are representative of the wide variation about these points. These results indicated that there was no statistically significant difference among the various configuration isomers in the rate at which they formed complexes (Table 1). In contrast, no matter which configuration isomer, the lack of complex formation in the dimer was obtained, which is consistent with the negligible reaction rate observed in the dimer. The ongoing simulations are focused on more challenging targets: tetramer and larger ring-size complexes.
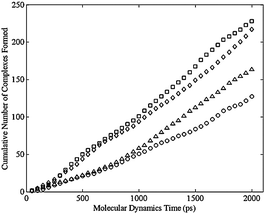 | ||
| Fig. 8 Cumulative number of bimetallic complexes formed per cobalt atom versus simulation time for the trimer, averaged over 40 simulation runs, for the cis-cis-cis (square), cis-cis-trans (diamond), cis-trans-trans (triangle), and trans-trans-trans (circle) isomers. | ||
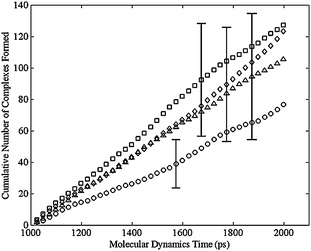 | ||
| Fig. 9 Cumulative number of bimetallic complexes formed per cobalt atom versus simulation time for the trimer, averaged over 40 simulation runs with the equilibration period of 1000 ps removed, for the cis-cis-cis (square), cis-cis-trans (diamond), cis-trans-trans (triangle), and trans-trans-trans (circle) isomers. The error bars show the 90% confidence interval for the 40 simulation runs at various simulation times. | ||
Catalytic reaction scope of the higher molecular weight catalysts
Since the larger ring-size Co(III)-salen complexes (tetramer-hexamer and pentamer-decamer) showed a significant improvement of catalytic activity for the HKR of allyl glycidyl ether in comparison to the original mixture, we evaluated the reaction scope of these oligomers using the structurally diverse terminal epoxides described in Table 2. To compare these higher molecular weight catalysts easily to the original mixture, we followed the original reaction conditions including catalyst loading, temperature and reagent amounts.20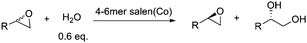
| Entry | R | Loadingb (mol%) | Time | eec (%) | Conversiond (%) |
|---|---|---|---|---|---|
| a Reactions were carried out with 0.05 mol epoxides under solvent-free conditions at 25 °C (except for entry 4). b Catalyst loading based on cobalt. For comparison, the same loadings as our previous reports were applied.20 c Determined by chiral GC or HPLC methods. d Determined by GC analysis with chlorobenzene as internal standard. e Resolutions were carried out at ambient temperature for 1 h and then heated to 40 °C. | |||||
| 1 | n-Bu | 0.01 | 80 min | >99 | 51.2 |
| 2 | CH2Cl | 0.01 | 100 min | >99 | 52.8 |
| 3 | CH2OAllyl | 0.01 | 5 h | >99 | 51.1 |
| 4e | CH2OPh | 0.01 | 2 h | >99 | 50.5 |
| 5 | Ph | 0.1 | 3.5 h | >99 | 50.7 |
| 6 | t-Bu | 0.25 | 5 h | >99 | 51.9 |
The results described in Table 2 show that at the same catalyst loading the tetramer-hexamer mixture can significantly shorten the reaction time compared to the original Co(III)-salen mixture.20 For instance, the original Co(III)-salen mixture took 120 min and 150 min to resolve 1,2-epoxyhexane and epichlorohydrin, respectively, while the tetramer-hexamer Co(III)-salen oligomers reduced the reaction time to 80 min for 1,2-epoxyhexane (Table 2, entry 1) and 100 min for epichlorohydrin (Table 2, entry 2). In the HKR of phenyl glycidyl ether, the tetramer-hexamer catalysts could dramatically shorten the reaction time from 20 h by the original mixture to 2 h (Table 2, entry 4). For some challenging HKR target molecules, like styrene oxide, a conjugated epoxide, and tert-butyloxirane, a substantial sterically hindered epoxide, the larger ring-size mixture exhibited superior catalytic activities. Racemic styrene oxide was resolved in 3.5 h in contrast to 24 h using the original mixture (Table 2, entry 5). The resolution of tert-butyloxirane was completed in 5 h (Table 2, entry 6). In comparison, it took 48 h for the original mixture to reach an ee of 98%. These experiments clearly demonstrated that the larger ring-size oligomers possess outstanding catalytic activities and enantioselectivities and also are featured with excellent functional group tolerance.
Asymmetric ring-opening of epoxides using other nucleophiles and an internal epoxide
After the successful HKR tests, we further explored the reaction scope of the tetramer-hexamer Co(III)-salen mixture. In the literature, the asymmetric ring-opening of epoxides with nucleophiles other than water has been challenging.19 We investigated whether our larger ring-size oligomers can catalyze a broad range of nucleophiles. First, we investigated the use of phenol as nucleophile to perform the asymmetric ring-opening of epichlorohydrin (Scheme 1A). A catalyst loading of 0.25 mol% completes the kinetic resolution in 4 h and (R)-1-chloro-3-phenoxy-2-propanol was produced in 99% ee and isolated in 98% yield. Trimethylsilyl ethanol, as an example of a primary alcohol, was then examined as nucleophile (Scheme 1B). The resolution of 1,2-epoxyhexene was catalyzed by 0.2 mol% catalyst and completed in 5 h. (R)-1-(2-Trimethylsilyl ethoxy)-2-hexanol was obtained as almost a single enantiomer in 96% isolated yield.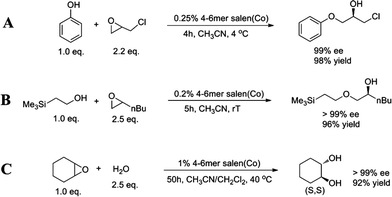 | ||
| Scheme 1 Asymmetric ring-opening reactions using the tetramer-hexamer Co(III)-salen mixture. (A) Asymmetric ring-opening of epichlorohydrin with phenol, (B) asymmetric ring-opening of 1,2-epoxyhexane with trimethylsilyl ethanol, and (C) asymmetric ring-opening of cyclohexene epoxide with water. | ||
Finally, we investigated a non-terminal epoxide, cyclohexene epoxide, which also has been shown to be problematic with other catalysts (Scheme 1C).45,46 Using the tetramer-hexamer Co(III)-salen mixture (1 mol% catalyst loading at 40 °C) the hydrolysis of cyclohexene epoxide took more than two days, providing 92% yield of (S,S)-trans-cyclohexane-1,2-diol. While water was added in excess (2.5 equivalence), the ee of the final product was >99%, which demonstrates again the outstanding enantioselectivity of the larger macrocyclic catalysts.
Conclusions
In this manuscript, we explore the effect of ring size of a cyclic oligomeric Co(III)-salen catalyst system on the catalytic activity and enantioselectivity. A purification protocol was established to separate the different ring-size oligomeric salen ligands resulting in the isolation of dimers, trimers, tetramers, a tetramers to hexamers mixture and a pentamers to decamers mixture. Using allyl glycidyl ether as a model substrate, we demonstrate that the metalated dimer has no catalytic activity and that catalyst activity dramatically increases with increasing ring size until larger size oligomers exhibited superior catalytic efficiencies. The cause of the different catalytic behavior was investigated by NMR experiments and computational modeling. It is suggested that the dimer ring is strained preventing cooperative interactions between pendent salen moieties, which results in the poor reactivity for the bimetallic mechanism based HKR reactions. For higher mass oligomers, the accelerated reaction rate is most plausibly contributed to the better flexibility of pendent Co(III)-salens afforded by the increased ring size, so the catalytic center can reach an ideal distance and/or space arrangement for HKR reactions. Furthermore, the larger the ring-size of the oligomeric catalyst systems, the more Co(III)-salen catalytic moieties in the same molecule are capable of being involved by either complexing the epoxide or delivering the nucleophile. This thermodynamic factor has been reported to play a role in the cooperative de-deuteration of aldehyde-2-d by PEI, which reaction rates are also second-order dependence on the catalytic species.28,47The catalytic ability of higher mass Co(III)-salen oligomers was further tested by HKR of various terminal epoxides with significant increase of the reaction rates in comparison to our original catalyst mixture. Furthermore, higher mass Co(III)-salen oligomers can also efficiently catalyze the asymmetric ring-opening of epoxides with phenol and alcohol with excellent enantiomeric excesses and yields. It has been reported that between the two most active HKR catalysts known to date, our macrocyclic mixture 3 exhibit a slightly higher catalytic activity than macrocyclic catalyst 2 under rigorous identical conditions (counterion, substrate, temperature, catalyst loading, reagent amounts and ratios are exactly same).24 In this study, the large ring-size macrocyclic catalysts (tetramer to hexamer and pentamer to decamer) demonstrate remarkable increased catalytic efficiencies and selectivities in comparison to the original mixture 3 under identical reaction conditions. Therefore, it stands to reason that the large ring-size macrocyclic catalysts obtained in this study are the most active and selective Co(III)-salen catalysts reported to date.
In addition to sieving out superior Co(III)-salen catalytic species for asymmetric ring-opening of epoxides, this study demonstrates the significance of ring size of cyclic oligomeric catalysts towards cooperative reactions. Ring size effects should be taken into consideration in the design of future macrocyclic Co(III)-salen complexes and other macrocyclic oligomeric catalysts. Future direction includes scale-up syntheses of the big ring-size oligomers by the solid phase peptide ring-closing reaction or other templated ring-closing reactions.
Acknowledgements
We are thankful to the Department of Energy Office Basic Energy Sciences through Catalysis Contract No. DEFG02-03ER15459 for financial support of this work. The MALDI instrument was purchased through a grant from the National Science Foundation (CHE-0958457).Notes and references
- R. A. Copeland, Enzymes. A practical introduction to structure, mechanism, and data analysis, John Wiley & Sons Inc., 2000 Search PubMed.
- J. W. A. Steed, J.L., Supramolecular Chemistry, John Wiley & Sons Inc., 2000 Search PubMed.
- M. Komiyama, Prog. Polym. Sci., 1993, 18, 871–898 CrossRef CAS.
- A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707–724 CrossRef CAS.
- J. K. M. Sanders, Chem.–Eur. J., 1998, 4, 1378–1383 CrossRef.
- F. Diederich, G. Schurmann and I. Chao, J. Org. Chem., 1988, 53, 2744–2757 CrossRef CAS.
- Z. Clyde-Watson, A. Vidal-Ferran, L. J. Twyman, C. J. Walter, D. W. J. McCallien, S. Fanni, N. Bampos, R. S. Wylie and J. K. M. Sanders, New J. Chem., 1998, 22, 493–502 RSC.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS.
- D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS.
- Y. M. Song, X. Q. Yao, H. L. Chen, C. M. Bai, X. Q. Hu and Z. Zheng, Tetrahedron Lett., 2002, 43, 6625–6627 CrossRef CAS.
- M. A. Kwon and G. J. Kim, Catal. Today, 2003, 87, 145–151 CrossRef CAS.
- M. Beigi, S. Roller, R. Haag and A. Liese, Eur. J. Org. Chem., 2008, 2135–2141 CrossRef CAS.
- X. L. Zheng, C. W. Jones and M. Weck, Adv. Synth. Catal., 2008, 350, 255–261 CrossRef CAS.
- B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309–1312 CrossRef CAS.
- R. Breinbauer and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 3604–3607 CrossRef CAS.
- J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687–2688 CrossRef CAS.
- X. L. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105–1112 CrossRef CAS.
- P. Goyal, X. L. Zheng and M. Weck, Adv. Synth. Catal., 2008, 350, 1816–1822 CrossRef CAS.
- X. F. Guo, Y. S. Kim and G. J. Kim, Top. Catal., 2009, 52, 153–160 CrossRef CAS.
- C. S. Gill, W. Long and C. W. Jones, Catal. Lett., 2009, 131, 425–431 CrossRef CAS.
- X. Zhu, K. Venkatasubbaiah, M. Weck and C. W. Jones, ChemCatChem, 2010, 2, 1252–1259 Search PubMed.
- J. M. Ready and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 1374–1377 CrossRef CAS.
- R. N. Loy and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 2786–2787 CrossRef CAS.
- D. E. White and E. N. Jacobsen, Tetrahedron: Asymmetry, 2003, 14, 3633–3638 CrossRef CAS.
- J. Hine and R. L. Flachska., J. Org. Chem., 1974, 39, 863–870 CrossRef CAS.
- J. M. Notestein and A. Katz, Chem.–Eur. J., 2006, 12, 3954–3965 CrossRef CAS.
- J. D. Bass, A. Solovyov, A. J. Pascall and A. Katz, J. Am. Chem. Soc., 2006, 128, 3737–3747 CrossRef CAS.
- N. Madhavan, C. W. Jones and M. Weck, Acc. Chem. Res., 2008, 41, 1153–1165 CrossRef CAS.
- S. Kemper, P. Hrobarik, M. Kaupp and N. E. Schloerer, J. Am. Chem. Soc., 2009, 131, 4172–4173 CrossRef CAS.
- H. Gunther and G. Jikeli, Chem. Rev., 1977, 77, 599–637 CrossRef.
- Interconversion between cis- and trans-isomers of cyclic monoene or diene have been thoroughly studied viaVariable Temperature NMR. The conversion temperatures range from room temperature to over 200 °C depending on the ring size, structure and modification. With the same ring size, the interconversion of cyclic diene is much easier than the corresponding cyclic monoene due to the lower racemization energy barrier. For a review on this discussion see: H. Gunther and G. Jikeli, Chem. Rev., 1977, 77, 599–637 Search PubMed and for the cyclic monoene and cyclic diene comparison see: C. B. Reese and A. Shaw, Chem. Commun., 1970, 1367–1368 CrossRef.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- S. H. Shi, L. Yan, Y. Yang, J. Fisher-Shaulsky and T. Thacher, J. Comput. Chem., 2003, 24, 1059–1076 CrossRef CAS.
- K. Malek, A. P. J. Jansen, C. Li and R. A. van Santen, J. Catal., 2007, 246, 127–135 CrossRef CAS.
- W. H. Leung, E. Y. Y. Chan, E. K. F. Chow, I. D. Williams and S. M. Peng, J. Chem. Soc., Dalton Trans., 1996, 1229–1236 RSC.
- J. J. Chapman, C. S. Day and M. E. Welker, Eur. J. Org. Chem., 2001, 2273–2282 CrossRef CAS.
- C. T. Cohen, C. M. Thomas, K. L. Peretti, E. B. Lobkovsky and G. W. Coates, Dalton Trans., 2006, 237–249 RSC.
- B. L. Holian, A. J. Degroot, W. G. Hoover and C. G. Hoover, Phys. Rev. A, 1990, 41, 4552–4553 CrossRef.
- The distance between two Co centers for a possible reaction has not been determined experimentally or theoretically. Here, 10 Å was chosen because it is approximately the separation of the catalyst sites due to the acetate counter ion commonly used in these catalyst systems. A sensitivity analysis of this separation distance showed that this distance was also reasonably sensitive to the occurrence of potential reaction events as a function of macrocyclic octane geometries.
- M. Wojciechowski and B. Lesyng, J. Phys. Chem. B, 2004, 108, 18368–18376 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Professional, 1998 Search PubMed.
- K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008–11009 CrossRef CAS.
- S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252–2260 CrossRef CAS.
- J. Hine and F. A. Via, J. Am. Chem. Soc., 1972, 94, 190–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of catalyst synthesis, separation and characterizations, the HKR reactions, the epoxide ring-opening reactions, and simulation parameters. See DOI: 10.1039/c0sc00517g |
| This journal is © The Royal Society of Chemistry 2011 |