Highly selective additions of hydride and organolithium nucleophiles to helical carbenium ions†
Joyram
Guin
a,
Céline
Besnard
b,
Philip
Pattison
cd and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, Quai Ernest Ansermet 30, Ch-1211, Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch; Fax: +41 22 379 3215
bLaboratoire de Cristallographie, University of Geneva, Quai Ernest Ansermet 24, CH-1211, Geneva 4, Switzerland
cLaboratoire de Cristallographie, EPFL, CH-1015, Lausanne, Switzerland
dSwiss-Norwegian Beamline, ESRF, BP-220 F-38043, Grenoble Cedex, France
First published on 3rd December 2010
Abstract
Unsymmetrical cationic [4]helicenes react with hydride or organolithium reagents and the two diastereotopic faces can be discriminated with high efficiency (dr up to and higher than 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)
:1)
Stereoselective additions of nucleophiles to trivalent carbon atoms are frequently-met reactions in organic chemistry and synthetic chemists often rely on stereogenic elements close to reactive trigonal centers to influence the stereochemical outcome of addition reactions.1,2 In the case of carbenium ions, recent studies have shown that neighboring tetrahedral stereogenic centers can control effectively the sense of induction.3,4 On these substrates bearing three different substituents around the positively charged atom and a stereocenter next to it (1, Fig. 1), nucleophiles react with essentially one of the two diastereotopic faces to yield addition products with high diastereoselectivity (dr ∼97
:3).5–10 In the present manuscript, the question of facial diastereoselectivity in addition reactions onto stabilized carbocations is also addressed, from a rather different perspective though. Using chiral carbenium ions of type 2 (Fig. 1 and 2), we demonstrate that a helical framework surrounding the electrophilic center can also be an effective stereocontrol element. Hydride and organolithium nucleophiles afford addition products with diastereoisomeric ratios up to and higher than 49:1.
 | ||
| Fig. 1 Facial diastereoselectivity in addition to carbenium ions with α-stereocenter and helicity as stereocontrol elements respectively. | ||
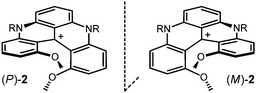 | ||
| Fig. 2 P and M enantiomers of cations 2. | ||
Previously, quinacridinium derivatives 2 have been reported.11,12 These compounds are readily prepared in one-step by reactions of primary amines with tris(2,6-dimethoxyphenyl)methylium ion 3 (Scheme 1). The reactions proceed through successive ortho SNAr reactions of methoxy substituents. Compounds 2 are highly stable carbocations (pKR+∼19)13 and, in terms of stereochemistry, they adopt a twisted helical conformation due to the strong steric repulsions occurring between the methoxy substituents in positions 1 and 13. Structurally, the two oxygen atoms are exactly above one another, and their mutual distance defines the inner pitch of the helicene system to be in the range of 2.7 Å. In case of the carbohelicenes, e.g.[6]helicene, pitches of 3.2–3.3 Å were found.14 Compounds 2 exist therefore as helical P and M enantiomers (Fig. 2) and are among the most configurationally stable helicenes.11,15 Of importance to this article, these carbocations react with nucleophiles: hydride and organometallic reagents in particular.15–18,19–23
![Stepwise synthesis of unsymmetrical [4]helicenes.](/image/article/2011/SC/c0sc00525h/c0sc00525h-s1.gif) | ||
| Scheme 1 Stepwise synthesis of unsymmetrical [4]helicenes. | ||
To perform the intended study on the facial selectivity of nucleophilic addition reactions,24 two different substituents were introduced on the nitrogen atoms (Fig. 3, R1 ≠ R2); the plane defined by the three neighboring substituents at the cationic carbon center exhibiting the diastereotopic faces. For R1, small, long and branched alkyl chains in the form of methyl (a), n-octyl (b), and isopropyl (c) groups were selected along with phenyl (d) and bulky mesityl (2,4,6-trimethylphenyl) (e) aromatic moieties. Precedents indicated that the corresponding alkyl and aromatic amines ought to react with cation 3 at room or slightly elevated (50 °C) temperatures and afford the corresponding acridinium derivatives 4 (Scheme 1).25 For R2, a different side-chain was required and it was known that an aliphatic amine such as nPrNH2 (25 equiv., 90 °C) ought to react with acridinium ions 4 to form the second nitrogen bridge.11
![Diastereoisomeric products resulting from additions on the re and si faces of [4]helicenes of type 2 represented with a M configuration. An arbitrary priority of R1 over R2 is considered.](/image/article/2011/SC/c0sc00525h/c0sc00525h-f3.gif) | ||
| Fig. 3 Diastereoisomeric products resulting from additions on the re and si faces of [4]helicenes of type 2 represented with a M configuration. An arbitrary priority of R1 over R2 is considered. | ||
Synthesis of acridinium salts [4a][BF4] to [4e][BF4] was achieved as planned (64–95%, see the supporting information†). These moieties reacted then with n-propylamine to afford the corresponding salts [2a][BF4] to [2e][BF4] (Scheme 1, 66–86%). X-ray quality crystals of [2e][BF4] were further obtained from an isopropanol solution (Fig. 4).† Cation 2e adopts a twisted helical conformation typical of this type of [4]helicene.26 No particular deformation was noticed from the presence of the bulky mesityl substituent; the two nitrogen atoms N1 and N2 (carrying substituents R1 and R2) are both sp2-hybridized.
![Ortep view of the crystal structure of [2e][BF4] (M enantiomer shown). One molecule of isopropanol and all hydrogen atoms are omitted for clarity. The BF4− anion is partially disordered.](/image/article/2011/SC/c0sc00525h/c0sc00525h-f4.gif) | ||
| Fig. 4 Ortep view of the crystal structure of [2e][BF4] (M enantiomer shown). One molecule of isopropanol and all hydrogen atoms are omitted for clarity. The BF4− anion is partially disordered. | ||
The study of the diastereoselectivity of the nucleophilic addition was started by first treating salts [2a][BF4] to [2e][BF4] with NaBH4 (2.0 equiv., EtOH, 20 °C). The cations underwent smooth reduction to afford derivatives of type 5 (Fig. 5). Within 30 min, the reaction mixtures changed from dark green suspensions to essentially colorless solutions indicating the completion of the additions. NMR spectroscopic analysis of the crude mixtures showed the presence of the reduced products 5 only, of which the ratios between the diastereoisomers could be readily determined by the integration of the respective signals.27 They are reported in Table 1 along with the yields of products isolated after a quick filtration over silica gel.
![Diastereoisomeric products resulting from additions on the re face of [4]helicenes of type 2 represented with a M configuration. For the re face assignment, a priority of R1 over Pr is considered.](/image/article/2011/SC/c0sc00525h/c0sc00525h-f5.gif) | ||
| Fig. 5 Diastereoisomeric products resulting from additions on the re face of [4]helicenes of type 2 represented with a M configuration. For the re face assignment, a priority of R1 over Pr is considered. | ||
For 5b, an essentially 1:1 mixture of diastereoisomers was obtained; this lack of selectivity being expected as linear n-Pr and n-Oct chains have similar conformations and provide little difference close to the reactive center. Otherwise, a preferred formation of one diastereoisomer was observed in all cases. For compounds 5a, 5c and 5d, moderate levels of discrimination were achieved (dr up to 7:1). However, one set of signals strongly predominated for compound 5e (dr 96:4); the improved selectivity being confirmed by HPLC analysis.
With this positive result, the chemistry was extended to other nucleophiles and aromatic moieties in particular. Salts [2a][BF4] to [2e][BF4] were treated with PhLi (3.0–4.0 equiv., THF, −78 °C). The results are reported in Table 2. In all cases, the reactions proceeded well and products 6a to 6e (Fig. 5) were isolated in good yields (86–98%). 1H NMR analysis of the crude mixtures demonstrated an effective facial discrimination of cation 2e and 2d as well (dr > 49:1, entries 4 and 5). Moderate levels of selectivity were again obtained with substrates 2a to 2c.
The continuation of the study, which consisted in broadening the nucleophile scope, was then conducted with salts [2d][BF4] and [2e][BF4] exclusively, because good selectivity seemed to be promoted only with an aromatic substituent as R1. The results are summarized in Table 3. Two alkyl- and one alkynyllithium nucleophiles were tested. For these reagents, a rather strong difference was noticed with salts [2d][BF4] and [2e][BF4]. Whereas low selectivity was achieved for the additions onto phenyl-substituted cation 2d (products 7d to 9d, entries 1 to 3), useful levels of facial discrimination (ratios ≥ 9:1) were obtained with bulkier cation 2e (products 7e to 9e, entries 6 to 8). In contrast, when aryllithium reagents were reacted, an excellent stereoselectivity (>49:1) was obtained in all cases irrespective of the nature of the nucleophile and of the cation (2d or 2e); the presence of electron-donating or withdrawing substituents at the para- or meta-positions of the incoming nucleophiles had no effect on the discrimination. On selected derivatives, HPLC analysis was performed which did not reveal the presence of a minor diastereoisomer. All in all, both salts displayed high diastereoselectivity in combination with ArLi reagents (products 6, 10 to 14) and discrimination with salt [2e][BF4] was effective even with hydride, alkyl- and alkynyllithium reagents.
| Entry | Cation | Added fragment | Product | Yield a | dr b |
|---|---|---|---|---|---|
| a Isolated yields, [%]. b Diastereoisomeric ratios determined by 1H NMR analysis of crude products. c Reactions were performed at 20 °C. | |||||
| 1 | 2d | CH3– | 7d | 93 | 63:37 |
| 2c | 2d | C6H5C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– C– |
8d | 88 | 58:42 |
| 3 | 2d | Me3SiCH2– | 9d | 92 | 56:44 |
| 4 | 2d | 4-CH3–C6H4– | 10d | 91 |
>98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 :2
|
| 5 | 2d | 4-CH3O–C6H4– | 11d | 90 |
>98:2
|
| 6 | 2e | CH3– | 7e | 95 | 90:10 |
| 7c | 2e | C6H5CC– |
8e | 90 | 91:9 |
| 8 | 2e | Me3SiCH2– | 9e | 92 | 93:7 |
| 9 | 2e | 4-CH3–C6H4– | 10e | 92 |
>98:2
|
| 10 | 2e | 4-CH3O–C6H4– | 11e | 90 |
>98:2
|
| 11 | 2e | 4-Br–C6H4– | 12e | 93 |
>98:2
|
| 12 | 2e | 3,5-(CH3)2–C6H4– | 13e | 93 |
>98:2
|
| 13 | 2e | 3,5-(CF3)2–C6H4– | 14e | 94 |
>98:2
|
Determination of the relative configuration of the diastereoisomers was performed by NMR spectroscopy28 and confirmed by X-ray structural analysis. X-ray quality crystals (Fig. 6) were afforded by slow evaporation of a solution (1:1 pentane:Et2O) of compound 14e, made by the addition of 3,5-(CF3)2–C6H4–Li to salt [2e][BF4] (dr > 49:1). As expected, the compound crystallized as a single diastereoisomer; the configuration of which being (M,R) or (P,S). The stereochemical descriptors M and P are kept in reference to the original helical conformations of the helicene precursor; R and S describe the configuration of the newly generated sp3 stereocenter. Stereoisomers (M,R) or (P,S) arise from a preferred addition of the ArLi nucleophile on the diastereotopic re face of the racemic helical cation.
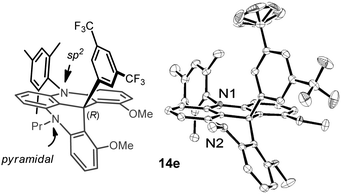 | ||
| Fig. 6 Drawing of 14e and Ortep view of its crystal structure (only the (M,R)-diastereoisomer shown). The (P,S)-stereoisomer, part of the propyl side chain and all hydrogen atoms are omitted for clarity. | ||
From the solid-state structure, further information was obtained. First, as a consequence of the change of hybridization of the central carbon from sp2 to sp3, the molecule does not adopt a strict helical conformation of the ortho-condensed framework anymore. On one side, three contiguous six-membered rings present an essentially planar arrangement in which the contained nitrogen atom is sp2 hybridized (here atom N1, Fig. 6). The arrangement of nitrogen N1 and the neighboring atoms is in fact quasi-planar with only a slight out-of-plane (OOP) distortion for N1 of 0.049 Å. On the other side, a strong bending of the backbone is enforced to compromise with the new carbon sp3 center and the repulsion of the methoxy substituents. This has consequences on the other nitrogen atom (N2, Fig. 6) which pyramidalizes to account for the folding of the corresponding six-membered ring that adopts an essentially boat-like conformation. The degree of the pyramidalization can be measured through the out-of-plane displacement of 0.227 Å of the N2-atom.
This geometrical distinction of the two nitrogen atoms (planar trigonal vs. pyramidalized) is probably the cause of the facial selectivity. The N-atom linked to the (bulky) aromatic substituent remains sp2-hybridized from the starting cation to the preferred diastereoisomeric product whereas the more flexible N-atom attached to the alkyl group bends to accommodate the strain induced by the developing nucleophilic attack. This geometrical constraint on the planar sp2N-Aryl atom and its surrounding rings then favors one main facial approach—the one that leads to the addition of the nucleophile to a lengthening of the distance between the MeO-substituents (Fig. 7, path a). The O⋯O distance in cation 1e and in adduct 14e is 2.673 and 2.754 Å, respectively. The other approach leads, on the contrary, to a highly disfavored tightening of the methoxy groups enforced by the rigidity (Fig. 7, path b). Consequently, for a cation 2d or 2e of M configuration, attacks on the diastereotopic re and si faces will result from the aromatic group being positioned as R1 and R2, respectively (Fig. 3). Opposite sense of induction will occur with a helicene of P configuration.
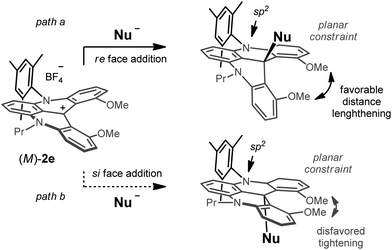 | ||
| Fig. 7 Geometrical constraint on the planar sp2N-Aryl atom and its surrounding aromatic groups as a possible origin for the diastereoselectivity. Path a: favored re face addition on (M)-2e (R1= mesityl). Path b: disfavored si face attack. | ||
In conclusion, high selectivity was achieved in nucleophilic attacks of hydride or organolithium reagents onto cationic [4]helicenes carrying aromatic groups on one nitrogen atom (dr up to and higher than 49:1). It shows that a helical framework surrounding an electrophilic center can be considered as an effective control element for the stereoselective addition of nucleophiles. In view of this result, it might now be interesting to study cations 2d and 2e as phase-transfer catalysts29 and see if their helical skeletons can help control selectivity on the nucleophile itself.
We thank Dr J. Mareda (Geneva) for helpful discussions. We are grateful for financial support of this work by the Swiss National Science Foundation, the State Secretariat for Education and Science for financial support.
Notes and references
- N. T. Anh, Top. Curr. Chem., 1980, 145–162.
- A. Mengel and O. Reiser, Chem. Rev., 1999, 99, 1191–1223 CrossRef CAS.
- P. G. Cozzi and F. Benfatti, Angew. Chem., Int. Ed., 2010, 49, 256–259 CAS.
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed.
- T. Ishikawa, T. Aikawa, Y. Mori and S. Saito, Org. Lett., 2004, 6, 1369–1372 CrossRef CAS.
- F. Mühlthau, O. Schuster and T. Bach, J. Am. Chem. Soc., 2005, 127, 9348–9349 CrossRef.
- F. Mühlthau, D. Stadler, A. Goeppert, G. A. Olah, G. K. S. Prakash and T. Bach, J. Am. Chem. Soc., 2006, 128, 9668–9675 CrossRef.
- P. Rubenbauer and T. Bach, Adv. Synth. Catal., 2008, 350, 1125–1130 CrossRef.
- D. Stadler and T. Bach, Chem.–Asian J., 2008, 3, 272–284 CrossRef CAS.
- D. Stadler, A. Goeppert, G. Rasul, G. A. Olah, G. K. S. Prakash and T. Bach, J. Org. Chem., 2008, 74, 312–318.
- B. W. Laursen and F. C. Krebs, Chem.–Eur. J., 2001, 7, 1773–1783 CrossRef CAS.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS.
- H. H. Freedman, in Reactive intermediates in organic chemistry, ed. G. A. Olah and P. v. R. Schleyer, Wiley-Interscience, New York, 1973, ch. 28 Search PubMed.
- K. P. Meurer and F. Vögtle, Top. Curr. Chem., 1985, 127, 1–76 CAS.
- C. Herse, D. Bas, F. C. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. W. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef.
- P. Mobian, N. Banerji, G. Bernardinelli and J. Lacour, Org. Biomol. Chem., 2006, 4, 224–231 RSC.
- B. Laleu, M. S. Machado and J. Lacour, Chem. Commun., 2006, 2786–2788 RSC.
- B. Laleu, P. Mobian, C. Herse, B. W. Laursen, G. Hopfgartner, G. Bernardinelli and J. Lacour, Angew. Chem., Int. Ed., 2005, 44, 1879–1883 CrossRef CAS.
- M. Lofthagen, R. Chadha and J. S. Siegel, J. Am. Chem. Soc., 1991, 113, 8785–8790 CrossRef CAS.
- M. Lofthagen, J. S. Siegel and M. Hackett, Tetrahedron, 1995, 51, 6195–6208 CrossRef CAS.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255–12263 CrossRef CAS.
- B. Baisch, D. Raffa, U. Jung, O. M. Magnussen, C. Nicolas, J. Lacour, J. Kubitschke and R. Herges, J. Am. Chem. Soc., 2009, 131, 442–443 CrossRef CAS.
- J. Guin, C. Besnard and J. Lacour, Org. Lett., 2010, 12, 1748–1751 CrossRef CAS.
- Prior to this study, addition reactions were only performed onto C2-symmetric substrates with identical nitrogen substituents (R1 = R2) and the plane defined by the three substituents at the cationic carbon center exhibited homotopic faces.
- F. C. Krebs, Tetrahedron Lett., 2003, 44, 17–21 CrossRef CAS.
- P. Mobian, C. Nicolas, E. Francotte, T. Bürgi and J. Lacour, J. Am. Chem. Soc., 2008, 130, 6507–6514 CrossRef CAS.
- In general, per compound, several signals are well split between the diastereoisomers. In the case of compounds 4, the singlet signal of the added hydrogen atom was particularly easy to monitor for the calculation of the diastereoselectivity.
- In compounds 6, and 10 to 14, the alkyl substituent on the pyramidalized nitrogen atom is strongly shielded by the added aromatic substituent and its protons appearing at lower frequency are an excellent signature.
- C. Nicolas and J. Lacour, Org. Lett., 2006, 8, 4343–4346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectral characterization of salts rac-2a-c, 2e and addition adducts 5a–e, 6a–e, 7d, 7e, 8d, 8e, 9d, 9e, 10d, 10e, 11d, 11e, 12e, 13e and 14e. Crystal structure determinations of [2e][BF4] CCDC 775297 and 14e CCDC 775298 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00525h |
| This journal is © The Royal Society of Chemistry 2011 |