Single microcrystals of organoplatinum(II) complexes with high charge-carrier mobility†
Chi-Ming
Che
*,
Cheuk-Fai
Chow
,
Mai-Yan
Yuen
,
V. A. L.
Roy‡
,
Wei
Lu
,
Yong
Chen
,
Stephen Sin-Yin
Chui
and
Nianyong
Zhu
Department of Chemistry, Institute of Molecular Functional Materials, HKU-CAS Joint Laboratory on New Materials, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China. E-mail: cmche@hku.hk
First published on 29th November 2010
Abstract
Hydrogen-bonding pyrazolyl and imidazolyl motifs are incorporated into organoplatinum(II) complexes and found to be harmonized with extended π–π and PtII⋯PtII interactions to align the planar cations into a quasi-1-D columnar structure or a quasi-2D framework. A field-effect electron mobility up to 20 cm2V−1s−1 has been recorded with a single-microcrystal transistor. In addition, crystalline samples of one of the organoplatinum(II) complexes show intriguing thermoluminescent and vapoluminescent properties.
Introduction
There is a surge of interest in developing semiconducting supermolecules and polymers for the realization of “plastic electronics”. Despite the advances made in this area, developing an organic semiconductor with high charge-carrier mobility, a prerequisite for the realization of high-performance organic devices and circuits with practical interest, remains a formidable challenge.Organic single crystals offer an excellent means to the exploration of charge-transporting properties as well as structure-property relationships. They are free from impurities and the long-range molecular arrangement in single crystals facilitates the charge-transporting processes, thus offering higher mobility and substantially better device performance when compared to their polycrystalline counterparts.1 There have been extensive studies revealing that planar aromatic organic molecules such as oligoacenes,2sulfur- and/or nitrogen-rich aromatic heterocycles,3porphyrins4 and phthalocyanines (Pcs)5 with intrinsic electron delocalization and intermolecular π–π interactions can direct anisotropic growth of molecular crystals with appreciable charge-carrier mobilities. On the contrary, related studies using single crystals of transition metal complexes are sparse.5,6 The versatile metal–ligand coordination and intermolecular metal⋯metal interactions are useful driving forces for molecular self-assembly and can be used to scaffold molecules into various superstructures. Among the transition metal complexes studied for this endeavor, organoplatinum(II) complexes have been attracting our attention. The square planar coordination geometry of PtII ions allows for the overlapping of 5dz2 orbitals in close proximity,7 and thereby providing a directional driving force for anisotropic growth of organometallic nanostructures.8 In recent works, we reported that supramolecular organoplatinum(II) nanowires can be adopted as semiconducting materials in an organic light-emitting thin-film field-effect transistor (OLEFET), albeit that the charge-carrier mobilities in that device are far from optimized.8d In the present work, we synthesized and structurally characterized a series of luminescent organoplatinum(II) complexes (1–6) with aromatic tridentate ligands containing hydrogen-bonding pyrazolyl9 and imidazolyl10 motifs. These planar molecules can be solution-processed into quasi-1D or -2D microcrystals exhibiting a hole mobility up to 1.8 cm2V−1s−1 and an electron mobility up to 20 cm2V−1s−1, as measured using a configuration of single-crystal field-effect transistor (FET). The charge-carrier mobilities of these organoplatinum(II) single microcrystals are among the highest when compared with that of the previously reported ambipolar organic single crystal semiconductors such as FePc, CuPc5 and tetracene.2d
Results and Discussion
Complexes 1–6 (Chart 1) were obtained by refluxing a methanolic solution of cis-[Pt(DMSO)2Cl2] and the corresponding ligand (see Supporting Information† for synthesis) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in the presence of HCl and under a nitrogen atmosphere. All of the platinum(II) complexes were isolated as chloride or perchlorate salts. The crystal structures of 1a·(CH3OH)2, 3a·(H2O)0.67, 3b, and 6·(DMSO)·(H2O)2 have been determined by X-ray crystallography.§The intermolecular interactions among and between the anions, solvated molecules and the hydrogen-bonding pyrazolyl and imidazolyl motifs play a key role in scaffolding the planar platinum(II)cations in a quasi-1D or -2D fashion.
:1 molar ratio in the presence of HCl and under a nitrogen atmosphere. All of the platinum(II) complexes were isolated as chloride or perchlorate salts. The crystal structures of 1a·(CH3OH)2, 3a·(H2O)0.67, 3b, and 6·(DMSO)·(H2O)2 have been determined by X-ray crystallography.§The intermolecular interactions among and between the anions, solvated molecules and the hydrogen-bonding pyrazolyl and imidazolyl motifs play a key role in scaffolding the planar platinum(II)cations in a quasi-1D or -2D fashion.
 | ||
| Chart 1 Chemical structures of complexes 1–6. | ||
The crystal structure of 1a reveals that the [Pt(H2L1)Cl]+ [H2L1 = 2,6-di(1H-pyrazol-3-yl)pyridine] cations (Fig. 1a) are oriented in head-to-tail pairs with extended intermolecular π–π and PtII⋯PtII interactions leading to a one-dimensional stacked structure (Fig. 1b) with alternating intermolecular Pt⋯Pt distances of 3.46 and 3.47 Å and a Pt⋯Pt⋯Pt angle of 162.65°. In addition, there are extensive intermolecular hydrogen-bonding interactions extended along the b-axis (see Supporting Information†). Each [Pt(H2L1)Cl]+ molecule is connected to two solvated methanol molecules through hydrogen-bonding between the Npyrazole–H and O atom of methanol (hydrogen-bonding distance = 1.865 Å). Similar columnar packing is also observed in crystal structures of 6 and 3b (see Supporting Information†), where the [Pt(L3)Cl]+ [L3 = 2,6-bis(1-methyl-1H-pyrazol-3-yl)pyridine] and [Pt(H2L6)Cl]+ [H2L6 = 2,6-di(1H-imidazol-2-yl)pyridine] cations are packed in a head-to-tail manner with interplanar distances of 3.17–3.40 Å but without short Pt⋯Pt distances (<3.5 Å). Similar to 1a, the Nimidazole–H moieties in complex 6 also participate in hydrogen-bonding network with solvent molecules. One of the N–H motifs on the tridentate ligand acts as hydrogen donor to the O atom of the solvated water molecule (hydrogen-bonding distance = 1.655 Å); while the other N–H motifs act as hydrogen donor to the O atom of the solvated DMSO molecule (hydrogen bonding distance = 2.309 Å). In both crystal structures of 1a and 6, the Npyrazole–H and Nimidazole–H motifs and solvated molecules form extensive hydrogen bonding networks at the periphery of the cationic columns.
![Perspective views of X-ray crystal structures of (a) 1a and (c) 3a. (b) Packing diagram of 1a viewed along c-axis shows one-dimensional PtII⋯PtII (dashed lines) and π–π stacking interactions. Packing diagrams of 3a viewed along (d) the [110] zone axis and (e) c-axis show two-dimensional π–π stacking interactions.](/image/article/2011/SC/c0sc00479k/c0sc00479k-f1.gif) | ||
| Fig. 1 Perspective views of X-ray crystal structures of (a) 1a and (c) 3a. (b) Packing diagram of 1a viewed along c-axis shows one-dimensional PtII⋯PtII (dashed lines) and π–π stacking interactions. Packing diagrams of 3a viewed along (d) the [110] zone axis and (e) c-axis show two-dimensional π–π stacking interactions. | ||
In the crystal structure of 3a (Fig. 1c), there are two sets of [Pt(L3)Cl]+ columnar chains which are arranged orthogonally to each other (Fig. 1d). In each column, the [Pt(L3)Cl] units are stacked in dimers and in head-to-tail fashion (torsional angle Npyridine–Pt1–Pt2–Npyridine = 171.28°) with π–π interactions (interplanar distance 3.25 to 3.55 Å) along the longitudinal axis of the column. When viewed along the c-axis, the two orthogonally arranged columns of [Pt(L3)Cl] cations display a two-dimensional network of π–π interactions (Fig. 1e). Notably, complex 3a ([Pt(L3)Cl]Cl) has the same cation and similar bonding parameters as 3b ([Pt(L3)Cl](ClO4), see Supporting Information†) and recently reported [Pt(L3)Cl](OTf),11 but the molecular arrangement and intermolecular interactions are remarkably altered upon changing the counter anion from chloride in 3a to perchlorate in 3b and to triflate in [Pt(L3)Cl](OTf). This finding signifies the pivotal role played by anions on the molecular packing of these organoplatinum(II) complex cations in the solid state.
The X-ray crystal structures reveal extensive intermolecular PtII⋯PtII and/or π–π interactions between the platinum(II) complexes, which can be harnessed to be the driving forces for the formation of nanostructures by self-assembly reactions. The morphologies of the nanostructures formed by precipitating DMF solutions (100 μL, 2 × 10−3 mol dm−3) into Et2O (2 mL) were examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). Fig. 2a and 2b show the TEM and SEM images of dispersed nanowires 1a with diameter of 35 ± 5.3 nm and length spanning over several micrometers (length 3.2 ± 0.8 μm). The selected area electron diffraction (SAED) pattern (Fig. 2c) of an individual nanowire revealed sharp diffraction spots with d-spacings ∼3.48 Å along the growth direction of the nanowire, corresponding to the [200] Miller planes of the single crystals of 1a, that is, the direction of the extended intermolecular π–π and PtII⋯PtII interactions. On the other hand, TEM and SEM images (Fig. 2d and 2e) revealed that the dispersion of 3a contains micrometer rectangular plates with both length and width of the plates in the range of 0.8–2 μm and thickness of 250–300 nm. Formation of plate-like morphology is presumably due to the presence of two-dimensional intermolecular π–π interactions as revealed from the single X-ray crystal structure of 3a. Interestingly, the SAED pattern of the submicron sized plates of 3a (Fig. 2f) showed two rows of diffraction spots of d-spacings of 3.34 and 3.49 Å in a orthogonal arrangement. These two d-spacings correspond to the [060] and [400] Miller planes, and are consistent with the presence of two-dimensional intermolecular π–π stacking interactions in the single crystal of 3a.
![SEM (a and d), TEM (b and e) images and SAED patterns (c and f) viewed along the [001] zone axis of self-assembled nanowires of 1a (a–c) and microplates of 3a (d–f).](/image/article/2011/SC/c0sc00479k/c0sc00479k-f2.gif) | ||
| Fig. 2 SEM (a and d), TEM (b and e) images and SAED patterns (c and f) viewed along the [001] zone axis of self-assembled nanowires of 1a (a–c) and microplates of 3a (d–f). | ||
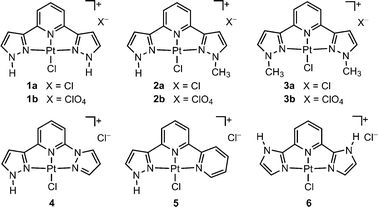
The organoplatinum(II) complexes display intriguing photophysical properties in solutions and in solid state. The electronic absorption spectra of complexes 1–6 in solutions at 298 K show intense vibronic-structured absorption bands at 250 to 350 nm (ε ∼ 104 dm3mol−1cm−1), a moderate intense absorption band at 350–400 nm (ε ∼ 103 dm3mol−1cm−1) and an absorption tail in the region > 420 nm (ε ∼ 102 dm3mol−1cm−1). The intense absorption bands at 250–350 nm are tentatively assigned to intraligand transitions (1IL) of the tridentate ligands. The lower energy absorption bands at 350–400 nm are assigned to spin-allowed 5d(Pt)→π*(L)1MLCT transition as both of its absorption wavelength and ε value are comparable to those of related transitions of [Pt(terpy)Cl]+ (terpy = 2,2′:6′,2′′-terpyridine),12 [Pt(C^N^N)Cl] (HC^N^N = 6-phenyl-2,2′-bipyridine)13 and [Pt(N^C^N)Cl] (N^CH^N = 1,3-bis(2′-pyridyl)benzene)14 complexes. Solvent effect on the electronic absorption spectrum was examined using 1a as an example. The low energy absorption band of 1a red-shifts from 356 nm with a tailing at 418 nm in DMF to 367 nm with a tailing at 424 nm in propan-2-ol (Fig. 3a). Upon excitation at 395 nm, 1a displays a vibronically structured emission band with λmax at 491 nm (τ = 2.90 μs; Φ = 0.063) in degassed methanol solution at room temperature (Fig. 3b). When the solvent changes from DMF to propan-2-ol, its emission maximum slightly red-shifts from 488 nm to 493 nm. The emission is tentatively assigned to mixed triplet metal-to-ligand charge transfer (3MLCT) and intraligand (3IL) excited states. In solid state at room temperature, all complexes are emissive (Fig. 3c) with broaden spectral shape and λmax generally red-shifted from their respective emission maxima recorded in solutions, revealing extensive intermolecular stacking interactions in solid state and coinciding with the X-ray and electron crystallographic studies.
 | ||
| Fig. 3 a) UV-vis absorption spectra of 1a in various solvents; b) emission spectra of 1a, 4, 5 in methanol and 6 in DMF solutions; and c) solid state emission of 1a, 3a, 4, 5, and 6 at 298 K. | ||
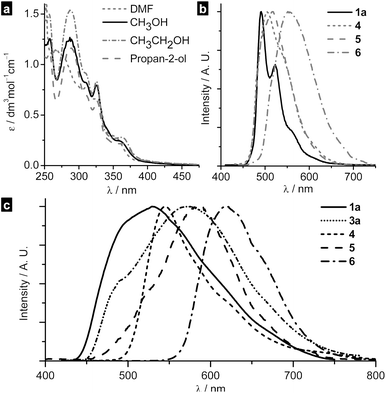
The charge transport properties of single microcrystals of platinum(II) complexes 1a, 3a, 3b, and 6 were examined using a field-effect transistor (FET) configuration. The dispersion of the single microcrystals was drop-cast onto an array of bottom-contact field-effect transistor with pre-fabricated source and drain electrodes. SiO2 was used as gate insulator and gold as source and drain electrodes. The field-effect mobilities thus obtained are summarized in Table 1. Fig. 4a shows an optical image of one of the devices and Fig. 4b and 4c show the n-channel and p-channel electrical characteristics of 1a, respectively. The drain-source current IDS increases with increasing both positive and negative gate voltages, indicating an ambipolar semiconducting behavior of crystal 1a. The channel was found to be open even at zero gate voltage, attributed to the bulk conduction of the crystal between the drain and source electrodes. Similar channel opening has been observed in previously reported organoplatinum(II) nanowires and polymers.8a,d,f,g Since saturation has not been attained for the field-effect transistor of 1a, the charge mobilities (μh and μe) were calculated at the linear regime (where VDS ≪ VG). The hole mobility and electron mobility of 1a are 0.4 cm2V−1s−1 (μh, at VG = −5 V) and 20 cm2V−1s−1 (μe, at VG = 5 V) respectively. Compared to previous reported mobility values of single-crystal field-effect transistors of FePc5a (μh = 0.3 cm2V−1s−1; μe = 0.03 cm2V−1s−1), CuPc5a (μh = 0.3 cm2V−1s−1; μe = 0.001 cm2V−1s−1), tetracene2f (μh = 0.16 cm2V−1s−1; μe = 0.037 cm2V−1s−1), rubrene2e (μh = 1.8 cm2V−1s−1; μe = 0.011 cm2V−1s−1) and tetraphenylpyrene2h (μh = 0.34 cm2V−1s−1; μe = 0.077 cm2V−1s−1), the charge carrier mobilities of 1a are the highest among the ambipolar single crystal field effect transistors reported in literature. Similar to 1a, all of the Pt(II) complexes examined in this work exhibited ambipolar output characteristics. The hole mobility and electron mobility of single crystals 3a, 3b, and 6 are in the range of 0.2–1.8 cm2V−1s−1 (μh) and 0.4–0.9 cm2V−1s−1 (μe), suggesting a balanced hole and electron transporting. The recorded hole mobilities of our single microcrystals are lower than that of dithiophene-tetrathiafulvalene single crystals (1.4 cm2V−1s−1)3a,b but comparable to that of triisopropylsilylethynyl pentacene (0.75 cm2V−1s−1).2g
| Crystal | Electron mobility μe/cm2V−1s−1 | Hole mobility μh/cm2V−1s−1 | PtII⋯PtII distance/Å | π–π distance/Å |
|---|---|---|---|---|
| a The charge-carrier mobility (μ) was calculated from the linear regime using IDvs.VG relation. In the linear regime where VDS ≪VGS, μ = (∂IDS/∂VGSVDS)(L/WC), where W is the channel width; L is the channel length; C is the capacitance of the SiO2 insulating layer; VGS is the gate voltage, VDS is the drain-source voltage, and IDS is the channel drain-source current. All the measurements were performed at room temperature under Ar atmosphere. | ||||
| 1a | 20 | 0.4 | 3.46, 3.47 | 3.41–3.45 |
| 3a | 0.5 | 0.2 | 3.65, 3.85 | 3.25–3.63 |
| 3b | 0.9 | 1.8 | 5.10, 6.82 | 3.14–3.43 |
| 6 | 0.4 | 0.6 | 4.33, 4.73 | 3.37–3.40 |
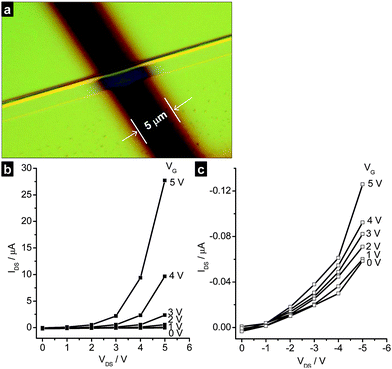 | ||
| Fig. 4 (a) Optical image of a single microcrystal of 1a fabricated into a field-effect transistor with a channel length of 5 μm. The p-channel (b) and n-channel (c) electrical characteristic (IDSvs.VDS) of single crystal 1a at various gate voltages. | ||
When discussing the operation mechanism of FET devices, we correlate the structural parameters with the high charge mobilities of the semiconducting microcrystals. (1) An important feature of our semiconducting crystals is their ionic nature. The cationic nature of the platinum(II) complexes, while biased in a field effect transistor structure, allows an electrical potential to be created at the metal/semiconductor interface. This facilitates electron and hole injection from the metal (source) to the semiconductor. (2) Once the charges are injected, charge hopping occurs through the molecules to eventually arrive at the drain electrode. In the present study, we found that extended hydrogen-bonding interactions in the crystal structures of 1a and 3a play a key role in scaffolding the planar platinum(II) cations in a quasi-1D columnar structure or a quasi-2D network. We propose that the orchestration of weak intermolecular PtII⋯PtII, π–π and/or hydrogen-bonding interactions facilitate long-range arrangement of square planar platinum(II) complexes into a cofacial configuration, which can enhance the charge-hopping process inside the single crystal. We reason that the chain-like metal–metal interactions provide an efficient pathway for electron transporting while the cofacial π–π interactions play a key role for hole transporting.
The X-ray crystal structure of 1a reveals continuous intermolecular PtII⋯PtII (3.46 Å) and π–π interactions (3.43 Å). The continuous PtII⋯PtII interactions of 1a bring the 5dz2 orbitals of the complex cations in close proximity. We propose that overlapping of the 5dz2 orbitals in a one-dimensional chain-like structure could generate a charge-hopping pathway accounting for the high electron mobility of 1a (20 cm2V−1s−1). However, the PtII⋯PtII distances within the crystal structures of 3a, 3b and 6 are 3.65, 5.10 and 4.33 Å respectively, which are too long for close PtII⋯PtII contacts (<3.5 Å). The absence of extended PtII⋯PtII interactions in the crystal structures of 3a, 3b and 6 is suggested to lead to relatively lower electron mobility values (0.5, 0.9 and 0.4 cm2V−1s−1 respectively).
On the other hand, the continuous π–π interactions in the crystal structures of 1a, 3a, 3b and 6 bring the p-orbitals of the tridentate π-conjugated ligands into close proximity. We propose that overlapping of the p-orbitals facilitates the hole transport within the crystal lattice. Intermolecular separation is an important parameter for charge-transporting processes.2–5 Generally the longer the distance, the lesser the charges that arrive at the opposite electrode. The average π–π distances are 3.43, 3.44, 3.29 and 3.38 Å for 1a, 3a, 3b and 6, respectively (3b < 6 < 1a and 3a). The increase in the average π–π distances could account for the decrease of the corresponding hole mobilities (0.4, 0.2, 1.8 and 0.6 cm2V−1s−1 for 1a, 3a, 3b and 6, respectively, i.e.3b > 6 > 1a and 3a).
It is noteworthy that the non-covalent intermolecular interactions found in the crystal structures of organoplatinum(II) complexes, especially the PtII⋯PtII contacts, are sensitive towards environmental variations.15 In a preliminary study, we have found that crystalline samples of complex 6 exhibited intriguing thermoluminescent (Fig. 5a,b) and vapoluminescent (Fig. 5c,d) properties. Briefly, desolvated crystals of complex 6 showed vibronically structured emission with λmax at 525 nm, whereas structureless emission with enhanced intensity and red-shifted energy (λmax > 620 nm) was recorded using these crystals upon increasing the temperature up to 50 °C or in contact with a variety of organic vapors. Stacked structure with close PtII⋯PtII and/or π–π contacts is obviously involved for the observed red emission.15 We thus envisage that multi-functionalized optoelectronic devices16 (such as transistor-based sensors) can be produced when the high field-effect carrier-mobility is combined with the environmental sensor capability of single crystals of organoplatinum(II) complexes.
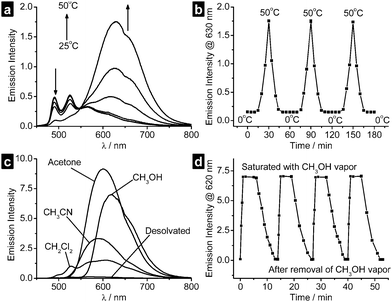 | ||
| Fig. 5 (a) Emission traces of a crystalline film of complex 6 in the temperature range of 25–50 °C. (b) A plot of emission intensity vs. time for such a film monitored at 630 nm, with temperature varying between 0 and 50 °C. (c) Emission spectra of a crystalline film of complex 6 in the desolvated state and in the presence of various organic vapors at 25 °C. (d) A plot of emission intensity vs. time for such a film monitored at 620 nm upon repeated saturation with methanol vapor and removal under vacuum. | ||
Acknowledgements
This work was supported by the University Development Fund of The University of Hong Kong, the University Grants Committee of the Hong Kong Special Administrative Region, China (Project No. [HKU 7008/09P]), and the CAS-Croucher Funding Scheme for Joint Laboratories. The work described in this paper was supported by a grant from the University Grants Committee of the Hong Kong Special Administrative Region, China (Project No. [AoE/P-03/08]).Notes and references
- (a) R. W. I. de Boer, M. E. Gershenson, A. F. Morpurgo and V. Podzorov, Phys. Status Solidi A, 2004, 201, 1302–1331 CrossRef CAS; (b) C. Reese and Z. Bao, Mater. Today, 2007, 10, 20–27 CrossRef CAS; (c) A. L. Briseno, S. C. B. Mannsfeld, M. M. Ling, S. Liu, R. J. Tseng, C. Reese, M. E. Roberts, Y. Yang, F. Wudl and Z. Bao, Nature, 2006, 444, 913–917 CrossRef CAS; (d) T. Hasegawa and J. Takeya, Sci. Technol. Adv. Mater., 2009, 10, 024314 CrossRef.
- (a) V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS; (b) A. N. Aleshin, J. Y. Lee, S. W. Chu, J. S. Kim and Y. W. Park, Appl. Phys. Lett., 2004, 84, 5383 CrossRef CAS; (c) L. B. Roberson, J. Kowalik, L. M. Tolbert, C. Kloc, R. Zeis, X. Chi, R. Fleming and C. Wilkins, J. Am. Chem. Soc., 2005, 127, 3069–3075 CrossRef CAS; (d) C. Reese, W.-J. Chung, M.-M. Ling, M. Roberts and Z. Bao, Appl. Phys. Lett., 2006, 89, 202108 CrossRef; (e) T. Takahashi, T. Takenobu, J. Takeya and Y. Iwasa, Appl. Phys. Lett., 2006, 88, 033505 CrossRef; (f) T. Takahashi, T. Takenobu, J. Takeya and Y. Iwasa, Adv. Funct. Mater., 2007, 17, 1623–1628 CrossRef CAS; (g) D. H. Kim, D. Y. Lee, H. S. Lee, W. H. Lee, Y. H. Kim, J. I. Han and K. Cho, Adv. Mater., 2007, 19, 678–682 CrossRef CAS; (h) S. Z. Bisri, T. Takahashi, T. Takenobu, M. Yahiro, C. Adachi and Y. Iwasa, Adv. Mater. Res., 2008, 10, 103–110 CAS.
- (a) M. Mas-Torrent, M. Durkut, P. Hadley, X. Ribas and C. Rovira, J. Am. Chem. Soc., 2004, 126, 984–985 CrossRef; (b) M. Mas-Torrent, P. Hadley, S. T. Bromley, X. Ribas, J. Tarrés, M. Mas, E. Molins, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2004, 126, 8546–8553 CrossRef CAS; (c) S. C. B. Mannsfeld, J. Locklin, C. Reese, M. E. Roberts, A. J. Lovinger and Z. Bao, Adv. Funct. Mater., 2007, 17, 1617–1622 CrossRef CAS; (d) O. D. Jurchescu, S. Subramanian, R. J. Kline, S. D. Hudson, J. E. Anthony, T. N. Jackson and D. J. Gundlach, Chem. Mater., 2008, 20, 6733–6737 CrossRef; (e) E. Ahmed, A. L. Briseno, Y. Xia and S. A. Jenekhe, J. Am. Chem. Soc., 2008, 130, 1118–1119 CrossRef CAS; (f) J. H. Oh, H. W. Lee, S. Mannsfeld, R. M. Stoltenberg, E. Jung, Y. W. Jin, J. M. Kim, J.-B. Yoo and Z. Bao, Proc. Natl. Acad. Sci. USA, 2009, 106, 6065–6070 CAS; (g) Z. Wei, W. Hong, H. Geng, C. Wang, Y. Liu, R. Li, W. Xu, Z. Shuai, W. Hu, Q. Wang and D. Zhu, Adv. Mater., 2010, 22, 2458–2462 CrossRef CAS.
- (a) T. Minari, M. Seto, T. Nemoto, S. Isoda, K. Tsukagoshi and Y. Aoyagi, Appl. Phys. Lett., 2007, 91, 123502 CrossRef; (b) P. Ma, Y. Chen, X. Cai, H. Wang, Y. Zhang, Y. Gao and J. Jiang, Synth. Met., 2010, 160, 510–515 CrossRef CAS.
- (a) R. W. I. de Boer, A. F. Stassen, M. F. Craciun, C. L. Mulder, A. Molinari, S. Rogge and A. F. Morpurgo, Appl. Phys. Lett., 2005, 86, 262109 CrossRef; (b) R. Zeis, T. Siegrist and C. Kloc, Appl. Phys. Lett., 2005, 86, 022103 CrossRef; (c) Q. Tang, H. Li, Y. Liu and W. Hu, J. Am. Chem. Soc., 2006, 128, 14634–14639 CrossRef CAS.
- K.-i. Sakai, T. Hasegawa, M. Ichikawa and Y. Taniguchi, Chem. Lett., 2006, 35, 302–303 CrossRef CAS.
- (a) T. W. Thomas and A. E. Underhill, Chem. Soc. Rev., 1972, 1, 99–120 RSC; (b) G. Gliemann and H. Yersin, Struct. Bonding, 1985, 62, 87–153 CAS; (c) D. M. Roundhill, H. B. Gray and C. M. Che, Acc. Chem. Res., 1989, 22, 55–61 CrossRef CAS; (d) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS.
- (a) W. Lu, V. A. L. Roy and C. M. Che, Chem. Commun., 2006, 3972–3974 RSC; (b) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J. P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659–2662 CrossRef CAS; (c) W. Lu, S. S. Y. Chui, K. M. Ng and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568–4572 CrossRef CAS; (d) M. Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M. H. So, S. S. Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895–9899 CrossRef CAS; (e) W. Lu, K. M. Ng and C. M. Che, Chem.–Asian J., 2009, 4, 830–834 CrossRef CAS; (f) W. Lu, Y. Chen, V. A. L. Roy, S. S. Y. Chui and C. M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621–7625 CrossRef CAS; (g) Y. Chen, K. Li, W. Lu, S. S. Y. Chui, C. W. Ma and C. M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS.
- C. K. Koo, Y. M. Ho, C. F. Chow, M. H. W. Lam, T. C. Lau and W. Y. Wong, Inorg. Chem., 2007, 46, 3603–3612 CrossRef CAS.
- B. S. Tovrog and R. S. Drago, J. Am. Chem. Soc., 1974, 96, 2743–2750 CrossRef CAS.
- L. Zhao, K. M.-C. Wong, B. Li, W. Li, N. Y. Zhu, L. X. Wu and V. W.-W. Yam, Chem.–Eur. J., 2010, 16, 6797–6809 CrossRef CAS.
- R. Bulchner, J. S. Field, R. J. Haines, C. T. Cunningham and D. R. McMillin, Inorg. Chem., 1997, 36, 3952–3956 CrossRef.
- T. C. Cheung, K. K. Cheung, S. M. Peng and C. M. Che, J. Chem. Soc., Dalton Trans., 1996, 1645–1651 RSC.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS.
- S. W. Lai and C. M. Che, Top. Curr. Chem., 2004, 241, 27–63 CAS.
- (a) Y. Kunugi, K. R. Mann, L. L. Miller and C. L. Exstrom, J. Am. Chem. Soc., 1998, 120, 589–590 CrossRef CAS; (b) M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nature, 2000, 406, 970–974 CrossRef CAS; (c) S. Diring, F. Camerel, B. Donnio, T. Dintzer, S. Toffanin, R. Capelli, M. Muccini and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 18177–18185 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, crystal packing diagrams, supplementary photophysical data and output characteristics of single-crystal field-effect transistor. CCDC reference numbers 763168–763170, 793196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00479k |
| ‡ Current address: Department of Physics and Materials Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong KongSAR, China. |
| § Crystal data. 1a·(CH3OH)2: C13H17Cl2N5O2Pt, M = 541.31, triclinic, a = 6.848(1), b = 11.736(2), c = 12.384(3) Å, α = 65.95(3)°, β = 79.94(3)°, γ = 76.84(3)°. V = 881.2(3) Å3, T = 301(2) K, space groupP − 1 (no. 2), Z = 2, 5150 reflections measured, 3268 unique (Rint = 0.0178) which were used in all calculations. The final wR(F2) was 0.0509 (all data). 3a·(H2O)0.67: C39H39Cl6N15O2Pt3, M = 1547.79, monoclinic, a = 12.588(2), b = 18.736(3), c = 20.805(4) Å, β = 106.39(2)°. V = 4707.3(1) Å3, T = 301(2) K, space groupP21/c, Z = 4, 27598 reflections measured, 8927 unique (Rint = 0.0524) which were used in all calculations. The final wR(F2) was 0.0693 (all data). 3b: C13H13Cl2N5O4Pt, M = 569.26, triclinic, a = 10.669(2), b = 10.411(2), c = 15.689(3) Å, β = 108.89(3)°. V = 1648.8(6) Å3, T = 301(2) K, space groupP21/c, Z = 4, 9437 reflections measured, 3116 unique (Rint = 0.0188) which were used in all calculations. The final wR(F2) was 0.0479 (all data). 6·(DMSO)·(H2O)2: C13H19Cl2N5O3PtS, M = 591.38, monoclinic, a = 9.4740(3), b = 7.1809(2), c = 27.9371(8) Å, β = 91.940(1)°. V = 1899.5(1) Å3, T = 173(2) K, space groupP21/c, Z = 4, 3299 reflections measured, 3123 unique (Rint = 0.048) which were used in all calculations. The final wR(F2) was 0.1200 (all data). |
| This journal is © The Royal Society of Chemistry 2011 |