Spectacular luminescent behaviour of tandem terpyridyl platinum(II) acetylide complexes attributed to solvent effect on ordering of excited states, “ion-pair” formation and molecular conformations†
Steven C. F.
Kui
,
Yuen-Chi
Law
,
Glenna So Ming
Tong
,
Wei
Lu
,
Mai-Yan
Yuen
and
Chi-Ming
Che
*
Department of Chemistry, Institute of Molecular Functional Materials, HKU-CAS Joint Laboratory on New Materials, State Key Laboratory on Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, SAR, China. E-mail: cmche@hku.hk; Fax: (+852) 2915 5176; Tel: (+852) 2859 7919
First published on 14th October 2010
Abstract
A series of oligomeric tandem terpyridyl platinum(II) complexes, namely [(tBu3tpy)Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ctpy)PtCl](OTf)2 (3), [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4), [(tBu3tpy)Pt(CCtpy)PtCCtpy](OTf)2 (5), and [(tBu3tpy)Pt(CCtpy)Pt(CCtpy)PtCl](OTf)3 (6), were prepared and their spectroscopic properties and self-aggregating behaviour were examined. In particular, complex 4 exhibits unusually higher emission quantum yield in CH2Cl2 (ϕ = 0.43) than that in CH3CN (ϕ < 0.1), which is attributed to the formation of a “contact ion pair” in chlorinated solvents, such as CHCl3, CH2Cl2 and C6H5Cl. DFT calculations revealed that both intersystem crossing (ISC) and radiative decay of this complex are less effective in CH3CN than in CH2Cl2, thus accounting for the low emission quantum yield in CH3CN.
Ctpy)PtCl](OTf)2 (3), [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4), [(tBu3tpy)Pt(CCtpy)PtCCtpy](OTf)2 (5), and [(tBu3tpy)Pt(CCtpy)Pt(CCtpy)PtCl](OTf)3 (6), were prepared and their spectroscopic properties and self-aggregating behaviour were examined. In particular, complex 4 exhibits unusually higher emission quantum yield in CH2Cl2 (ϕ = 0.43) than that in CH3CN (ϕ < 0.1), which is attributed to the formation of a “contact ion pair” in chlorinated solvents, such as CHCl3, CH2Cl2 and C6H5Cl. DFT calculations revealed that both intersystem crossing (ISC) and radiative decay of this complex are less effective in CH3CN than in CH2Cl2, thus accounting for the low emission quantum yield in CH3CN.
Luminescent platinum(II) complexes are distinctly different from octahedral d6 metal complexes of Ru(II) and Ir(III) in that the square planar geometry of Pt(II) complexes allows inner-sphere type metal–metal and metal–ligand interactions, which can profoundly impact the properties of electronic excited states. In this regard, platinum(II) complexes bearing π-conjugated N- and C-donor ligands were found to display intriguing photo-physical and luminescent properties1–8 that can be harnessed for practical applications in material science and luminescent sensory devices. A particular class of complexes possess a [(tpy)PtC
CAr]+ (tpy = 2,2′:6′,2′′-terpyridine; Ar = aryl group) motif which displays interesting aggregation behaviour as a consequence of intermolecular PtII⋯PtII and π⋯π interactions. The intermolecular PtII⋯PtII interactions have led to unique solvatochromism,5a,5c,9 temperature-dependent phosphorescence10 and formation of self-assembled gel11 and nanowires.12
In this work, the assembly of oligomeric Pt(II) complexes, [(tBu3tpy)Pt(CCtpy)PtCl](OTf)2 (3), [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4), [(tBu3tpy)Pt(CCtpy)PtCCtpy](OTf)2 (5), and [(tBu3tpy)Pt(CCtpy)Pt(CCtpy)PtCl](OTf)3 (6), all having {(tpy)PtCC} moieties tethered by a CC group, were prepared and their spectroscopic/photophysical properties were examined (Scheme 1).‡ Complex 2, used for the synthesis of 3–6, was first reported by Ziessel and co-workers.17,18 Notably, both high emission quantum yield (0.43) and long emission lifetime (11 μs) of [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4) in CH2Cl2 solution at room temperature are unique among the [(tpy)Pt(CCR)]+ complexes reported in the literature. This complex is weakly emissive in CH3CN solution, and its emission intensity in CH3CN is enhanced by adding chlorinated organic solvents (such as CHCl3, CH2Cl2, 1,2-Cl2C2H4, and C6H5Cl).
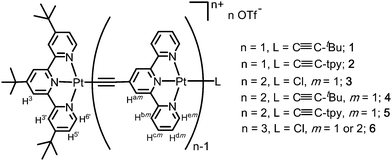 | ||
| Scheme 1 Platinum(II) complexes 1–6 studied in this work and the labelling scheme of aryl protons of the terpyridyl ligands with m = 1, 2 standing for the 1st and 2nd sets of acetylide terpyridyl, respectively. | ||
Based on 1H NMR and emission measurements, the luminophore cations of 4 were found to aggregate in CH3CN solution and such aggregation is suppressed in chlorinated solvents, which is attributed to the formation of a “contact ion pair”. The DFT calculations suggested a spectacular solvent effect on the ordering of excited states: the T2 is only 173 cm−1 above S1 in CH2Cl2, while ΔE(T3 − S1) = 426 cm−1 in CH3CN, and thus intersystem crossing (ISC) is more efficient in CH2Cl2. In addition, there is a low-lying triplet excited state, T2, in CH3CN which is only ∼500 cm−1 below the T3 state. Fast internal conversion from T3 to T2 state is thus possible. However, the Pt centre in this T2 state is different from that in the S1 state, rendering the direct spin–orbit coupling between T2 and S1 states ineffective. As such, harnessing the triplet excited state in CH3CN is inefficient, thus further contributing to its low emission quantum yield.
The method of Sonogashira13 was adopted for the synthesis of complexes 1, 2, 4, and 5, involving cross-coupling reactions of [(tBu3tpy)PtCl]+ with terminal acetylides, 4′-ethynyl-2,2′:6′,2′′-terpyridine (HCCtpy) or HCCtBu in the presence of diisopropylamine/copper iodide in CH2Cl2 or CH2Cl2–CH3CN. Refluxing [(COD)PtCl2] with 2 or 5 in H2O/acetone (1/4, v/v) gave 3 and 6 respectively. Results of 1H NMR, ESI-MS, FAB-MS studies and elemental analyses are consistent with the formulation of 1–6.
The labelling scheme for the aryl protons of 1–6 is shown in Scheme 1; the signals in the aromatic region (7.0–9.5 ppm) were assigned using 1H–1H COSY. The 1H NMR spectra of 3–6 are poorly resolved with broad peaks at ambient temperature and vary with temperature. As 4 has a better solubility than 3, 5 and 6 in CD3CN, 1H NMR study of 4 in CD3CN was performed over a larger temperature range (253 to 323 K; Fig. 1a) and at various complex concentrations (5.0 × 10−3 to 3.4 × 10−4 mol dm−3, Fig. 1b). Except for H3 and Ha1, all aryl proton signals shift downfield (0.11–0.30 ppm) as the temperature increases from 253 K to 323 K. At temperatures ≤ 298 K, the 1H NMR spectrum of 4 is poorly resolved with broad signals in the aromatic region. When the concentration of 4 is decreased from 5.0 × 10−3 to 3.4 × 10−4 mol dm−3, there is a gradual downfield shift of all aryl proton signals.
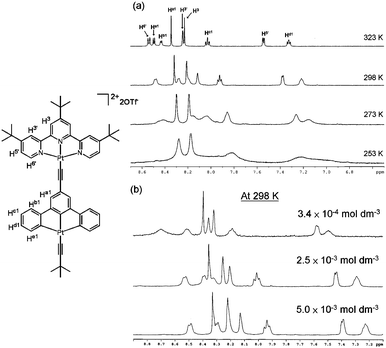 | ||
| Fig. 1 (a) Variable temperature 1H NMR spectra (500 MHz) of 4 in CD3CN solution (∼6 × 10−3 mol dm−3; 253–323 K). (b) 1H NMR spectra (400 MHz, 298 K) of 4 in CD3CN solutions at various concentrations: 3.4 × 10−4 (top); 2.5 × 10−3 (middle); 5.0 × 10−3 (bottom) mol dm−3. | ||
The 1H NMR spectra of 3 in DMF-d7 solution recorded at temperature from 299 to 393 K reveal a downfield shift of the aryl proton signals with increasing temperature (Fig. 2). The protons with the largest shift are those in close proximity to the platinum atom (H5′, H6′, Hd1 and He1, 0.52–0.81 ppm, from 299 to 393 K). The chemical shifts of H3, H3′, Ha1, Hb1 and Hc1 are less sensitive to temperature. This kind of behavior has similarly been observed in the 1H NMR spectrum of 5 in DMF-d7 solution at temperature from 300 to 393 K (Fig. S5).
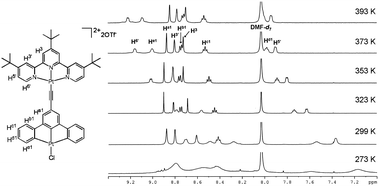 | ||
| Fig. 2 1H NMR spectra (500 MHz) of 3 in DMF-d7 solution at temperature 273–393 K. | ||
Broadening of the aryl proton signals at room temperature could originate from aggregation of the complexes in solutions. The aggregation behaviour of 3–6 as revealed by their 1H NMR spectra at temperature above 298 K is reminiscent of [(4-nBu–C6H4CC)Pt(tpy–R–tpy)Pt(CC–C6H4–nBu-4)](PF6)2 (R = O(CH2CH2O)3, O(CH2CH2O)4 or O(CH2)10O) reported by Yam and coworkers, who ascribed the line broadening and upfield shifting of the 1H NMR signals to self-assembly driven by intramolecular Pt⋯Pt and π⋯π stacking interactions.10 Here, we attribute the broad aryl proton signals of 3–6 at ambient temperature to intermolecular aggregation. Intramolecular Pt⋯Pt interaction would not be possible due to the short and rigid CC linkage in complexes 3–6; the decrease of rotational freedom of the aryl rings could only be accounted for by intermolecular Pt⋯Pt and/or π⋯π interactions. These intermolecular interactions increase the relaxation time of the proton signals which become chemically indistinguishable at ambient temperature. With an increase in temperature, aggregation is less favorable10 and the aryl proton signals of 3–6 become well-resolved. The downfield shifts of the aryl proton signals of 3–6 with increasing temperature are attributed to de-shielding of the aryl protons accompanied by disruption of the π⋯π stacking interactions. Consistent with the presence of molecular aggregation, the 1H NMR signals of 4 shift downfield with decrease in complex concentration since molecular aggregation is ruptured.
The absorption bands (Fig. S7) of 1–6 in CH3CN solutions at λmax < 400 nm are similar to the intraligand transitions (π→π*) of the tpy and acetylide ligands.3–7 With reference to previous studies on [(tpy)PtR]+ (R = acetylide),16 the low energy absorption peak(s) of the alkyl acetylide complexes 1 and 4 at λmax = 414 and 462 nm respectively are assigned to mixed metal-to-ligand (MLCT) and ligand-to-ligand charge-transfer (LLCT) transitions, 1[π(Pt–CC)→π*(tpy)]. The absorption bands of the aryl acetylide complexes 2 and 5 at λmax = 405 and 459 nm respectively, on the other hand, are assigned to 1LLCT (1[π(CCAr)→π*(tpy)]) transitions. For 3 and 6, with chloride directly attached to the platinum ion, their peak maxima at λmax = 423–426 nm are of 1MLCT character (1[dπ(Pt)→π*(tpy)]). Similarly, in CH3CN solution, the emission bands (Fig. S7) of 1 and 4 at λmax = 537 and 570 nm respectively are attributed to mixed excited states with 3MLCT/3LLCT character. The emissions of 3 and 6 (λmax = 544–546 nm) are attributed to 3MLCT excited states. For 2 and 5, the emission maxima at 519 and 549 nm in CH3CN are assigned to 3LLCT transitions.16 The emission spectrum of 5 is solvent dependent with λmax = 560 nm in CH2Cl2 and λmax = 550 and 671 nm in CH3CN (Fig. S7). This reveals that excimeric interaction (λmax = 671 nm) is more pronounced in CH3CN than in CH2Cl2 solutions. We propose that in CH3CN solutions, the [(tBu3tpy)Pt(CCtpy)PtCCtpy]2+ and TfO− ions are well separated and stacking interactions between {(tpy)Pt} moieties become more favourable. Interestingly, the intensity of the excimeric emission at λmax = 671 nm is more enhanced for 6, revealing that the {Pt(tpy)} moiety facilitates excimeric emission arising from π⋯π interactions between the neighbouring terpyridyl ligands.
Among the complexes [(tBu3tpy)Pt((CCtpy)Pt)n−1L]n+, we observed that the emission quantum yield of [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4) is solvent-dependent. This complex is strongly emissive in chlorinated solvents such as CHCl3 (ϕ = 0.39), CH2Cl2 (ϕ = 0.43) and 1,2-C2H4Cl2 (ϕ = 0.33); however it is weakly emissive/non-emissive in CH3CN, acetone, DMF, CH3OH and THF (Fig. 3).
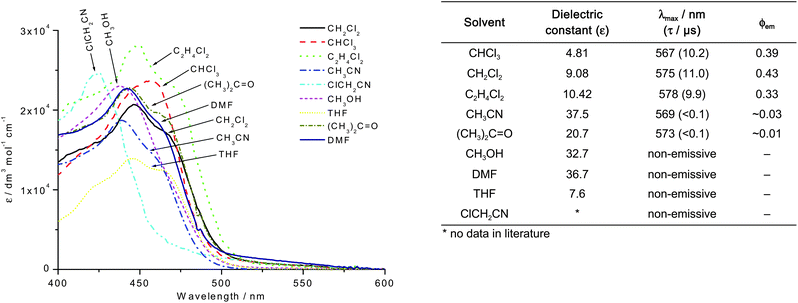 | ||
| Fig. 3 Left: UV-vis spectra of complex 4 in various organic solvents (concentration = 1 × 10−5 mol dm−3). Right: Table to show the emission data of complex 4 in various organic solvents. | ||
The UV-vis absorption and emission spectral changes of complex 4 in CH3CN mixed with CH2Cl2 at different compositions are depicted in Fig. 4. Upon increasing the CH2Cl2 composition while maintaining the complex concentration as constant (concentration = 2 × 10−5 mol dm−3), the absorption band at 438 nm (100% CH3CN) gradually shifts to 447 nm (100% CH2Cl2). At the same time, the emission quantum yield gradually increases from ϕ = 0.03 (100% CH3CN) to ϕ = 0.43 (100% CH2Cl2) and the emission maximum of 569 nm in 100% CH3CN is slightly shifted to 575 nm in 100% CH2Cl2.
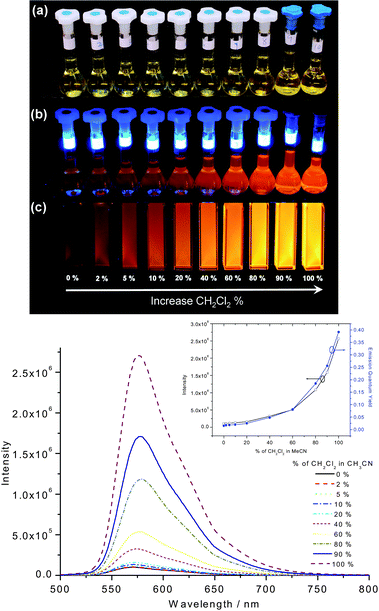 | ||
| Fig. 4 Upper: Photographs of complex 4 (concentration = 2 × 10−5 mol dm−3) in CH3CN mixed with 0–100% CH2Cl2 (a) under ambient light (in air), (b) under UV light irradiation (350 nm; in air), (c) under UV light irradiation (350 nm; in degassed environment). Bottom: Emission spectra of complex 4 (concentration = 2 × 10−5 mol dm−3) in CH3CN mixed with 0–100% CH2Cl2 (Inset: plots of intensity at 570 nm and emission quantum yield versus % of CH2Cl2 in CH3CN). | ||
Complex 4 is slightly soluble in chlorobenzene. Similar UV-vis absorption and emission spectral changes are observed upon mixing CH3CN solution of 4 with chlorobenzene at different compositions (0–90%; Fig. S8).We found that 4 in 10% CH3CN/90% chlorobenzene mixture shows an emission maximum at 575 nm with high emission quantum yield of 0.29. A series of organic solvents were added to CH3CN solution of 4 in order to examine the solvent effect on the emission intensity. The emission intensity of 4 in CH3CN (2 × 10−5 mol dm−3; 2 mL) is dramatically enhanced upon mixing with chlorinated organic solvents (1 mL), such as CH2Cl2, CHCl3, 1,2-Cl2C2H4, 1,1-Cl2C2H4, 1,1,2-Cl3C2H3, cis/trans-1,2-Cl2C2CH2, ClC6H5, 1,2-Cl2C6H4, 1,3-Cl2C6H4 or 1,3,4-Cl3C6H3. However, no detectable change of the emission intensity was found upon addition of the following solvents: CHBr3, CH3I, 1,2-Br2C2H4, C6H6, CH3CN, CH3OH, C2H5OH, CH3COOC2H5, (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C2H5OC2H5, DMF, H2O, 1,4-dioxane and THF to an CH3CN solution of 4.
O, C2H5OC2H5, DMF, H2O, 1,4-dioxane and THF to an CH3CN solution of 4.
In the literature, it has been reported that intermolecular or nanoscopic aggregation would induce enhancement in emission intensity but with little effect on the emission energy.14 In this work, no nanosized particles were observed by either dynamic light scattering (DLS) or transmission electron microscopy (TEM) measurements in pure CH2Cl2 and CHCl3 solutions of 4 or in CH3CN/ClC6H5 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9), CH3CN–CHCl3 (1:9) solution mixtures of 4, revealing that the emission enhancement is different from the systems reported in the literature.14 We propose that for this tandem terpyridyl platinum(II) acetylide complex [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4), solvent can affect (1) the formation of “ion pairs”15 and the molecular aggregation behavior and (2) the ordering of excited states. These two factors could contribute to the spectacular phenomenon of switching-on luminescence upon varying the solvent, and are discussed in the following sections.
:9), CH3CN–CHCl3 (1:9) solution mixtures of 4, revealing that the emission enhancement is different from the systems reported in the literature.14 We propose that for this tandem terpyridyl platinum(II) acetylide complex [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4), solvent can affect (1) the formation of “ion pairs”15 and the molecular aggregation behavior and (2) the ordering of excited states. These two factors could contribute to the spectacular phenomenon of switching-on luminescence upon varying the solvent, and are discussed in the following sections.
(1) Formation of “ion pairs” and molecular aggregation behavior
Since the solubility of a complex is mainly dependent on the solvent polarity, solvents of high polarity (dielectric constant εr > 20) dissolve ionic compounds to give solutions which are called “electrolytic solvents”. Solvents of low polarity (dielectric constant εr < 20) which dissolve ionic compounds poorly are termed “non-electrolytic solvents”.15 When the charged complex 4 is dissolved in chlorinated solvents such as CH2Cl2 and CHCl3 having low dielectric constants (εr < 10: for CH2Cl2εr = 9.1; for CHCl3εr = 4.8), formation of free complex cations becomes less favourable and “contact ion pairs” may result. Thus the intramolecular motion between two {(tpy)PtCC} moieties of complex 4 would be restricted by the counterion (Fig. 5). This will diminish the interactions between solvent molecules and the luminophore cation or two TfO− anions (Fig. 5).
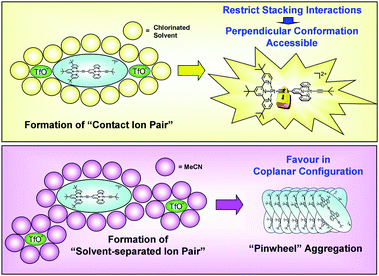 | ||
| Fig. 5 Upper: Formation of “contact ion pair” in chlorinated solvents (εr < 10). Bottom: Formation of “solvent-separated ion pair” in CH3CN (εr = 37.5). | ||
In solvents of intermediate dielectric constants (10 < εr < 40; such as CH3CN, εr = 37.5), ion pairs with solvent molecules surrounding the luminophore cation and two TfO− anions can be described as “solvent-separated ion pairs” (Fig. 5). In CH3CN, the π-conjugated luminophore cations of 4 would aggregate due to well isolated ions in “solvent-separated ion pairs”. By using a scanning electron microscope (SEM), slow evaporation of an CH3CN solution (∼ 2 × 10−4 mol dm−3) of complex 4 was found to give “donut” shaped submicron superstructures with diameters of 200–400 nm and with the average inner diameter of 60 nm. However an amorphous solid was obtained upon evaporation of CH2Cl2 solution (∼ 2 × 10−4 mol dm−3) of complex 4 (Fig. 6). This is attributed to the sterically bulky tert-butyl groups attached on both the terpyridine and acetylene ligands, which prohibit the head-to-head or head-to-tail stacking of the aryl units in CH3CN solution. We propose that the “donut” shaped nanostructure of complex 4 comes from the self-assembly of cations of 4 having “pinwheel” type aggregation with respect to each other to maximize π⋯π stacking and/or Pt⋯Pt interaction.
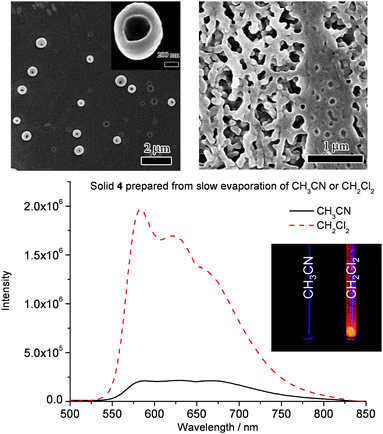 | ||
| Fig. 6 Upper: SEM images of complex 4 prepared from slow evaporation of CH3CN (left) and CH2Cl2 solution (right). Bottom: Emission spectra of solid 4 prepared from slow evaporation of CH3CN and CH2Cl2 (Inset: Photograph of different solid 4 under 350 nm UV light irradiation). | ||
To further account for the different aggregation behaviour in various solvents, we performed a series of 1H NMR and emission measurements in CD3CN and CDCl3/CD3CN mixtures at various volumic fractions of CDCl3. All of these experiments were carried out at controlled temperatures (298 K or 323 K) and a constant complex concentration (Fig. 7).
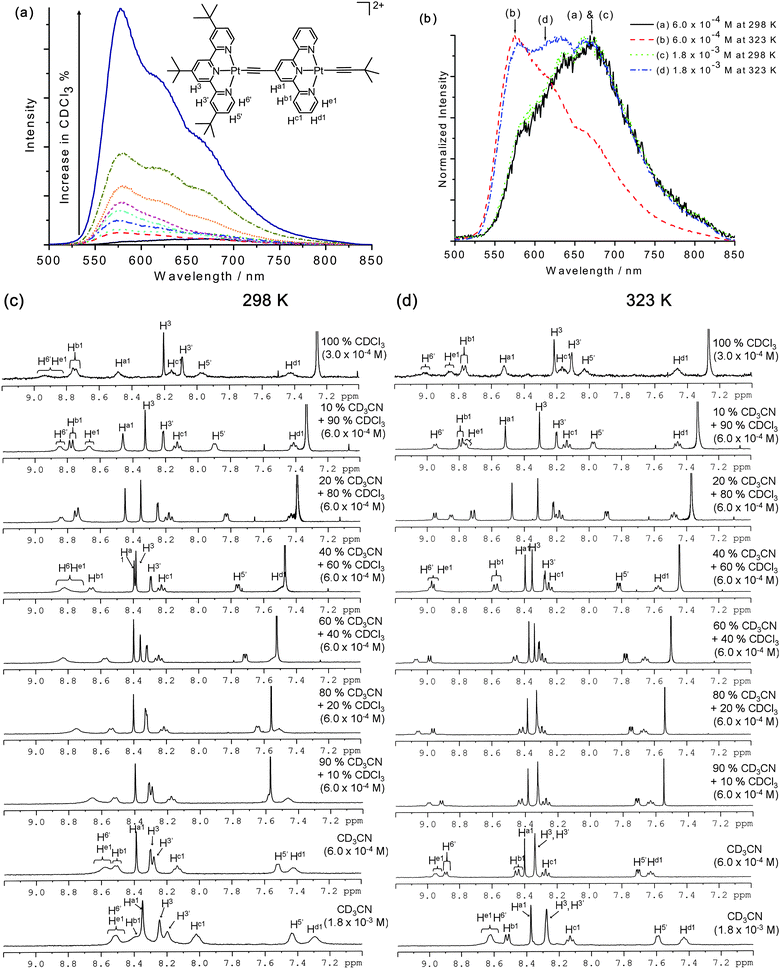 | ||
| Fig. 7 (a) Emission spectra of complex 4 in different compositions of CDCl3 (10% to 90%) in CD3CN mixture at 298 K. (b) Normalized emission spectra of complex 4 in CD3CN (concentration = 6.0 × 10−4 mol dm−3 and 1.8 × 10−3 mol dm−3) at 298 K and 323 K, respectively. (c) 1H NMR spectra at 298 K. (d) 1H NMR spectra at 323 K in CD3CN, CDCl3 and different compositions of CDCl3 (10% to 90%) in CD3CN mixture. | ||
At 298 K and at complex concentrations of 6.0 × 10−4 mol dm−3 and 1.8 × 10−3 mol dm−3 in CD3CN, the 1H NMR spectra are poorly resolved with broad signals in the aromatic region (Fig. 7c). Under these conditions, the emission spectra of these solutions reveal a weakly emissive band at λmax = 670 nm consistent with the proposition that cations of 4 aggregate in CD3CN solution (Fig. 7b) leading to broadening of 1H NMR signals. However, upon increasing the temperature to 323 K, the 1H NMR signals of 4 at 6.0 × 10−4 mol dm−3 become better-resolved (Fig. 7d) and the emission maximum is blue shifted to 577 nm though the emission intensity is still low. This suggests that the fast intermolecular motion of the CD3CN molecules and cations of 4 at higher temperature (323 K) would partially suppress the aggregation. On the other hand, the 1H NMR spectrum of 4 at 1.8 × 10−3 mol dm−3 exhibits broadened signals and the corresponding emission spectrum (Fig. 7b) shows a broad emission band with λmax from 577 nm to 670 nm revealing that the aggregation of cations of 4 in CD3CN still prevails at this complex concentration and temperature (323 K).
The 1H NMR signals of 4 become well-resolved and its emission intensity at 577 nm is enhanced, as the composition of CDCl3 increases from 10% to 90% in CDCl3/CD3CN mixture (6.0 × 10−4 mol dm−3) at both 298 K and 323 K (Fig. 7c and 7d). We suggest as the composition of CDCl3 increases the concentration of “contact ion pairs” and the degree of aggregation are suppressed. Subsequent restriction of intramolecular motion between two {(tpy)PtCC} moieties would enhance the radiative decay leading to an increase in emission intensity of 4 at 577 nm.
This explanation can be used to account for the strongly emissive properties of complex 4 (λmax = 576 nm; ϕ = 0.43) in CH2Cl2. The “contact ion pairs” of 4 in CH2Cl2 render the luminophore cations well-isolated from each other, thus disfavouring the intermolecular and intramolecular interactions accounting for high emission quantum yield. Indeed, this luminescent enhancement property has only been found in chlorinated solvents (except CCl4). Apart from CH3CN, this property has not been observed in solvents having dielectric constants in the range 10 < εr < 40 (such as DMF, DMSO, pyridine, CH3NO2, acetone, CH3OH, CH3CH2OH, etc.) and the solvents with low εr values such as Et2O, THF, dioxane, etc.
(2) Excited state ordering in various solvents
DFT calculations were performed on the model complex [((CH3)3tpy)Pt(CCtpy)PtCCCH3]2+ (4′), in which the tBu groups were replaced by CH3 groups to provide similar electronic effect but the computations were less demanding (Figure 8).
![Model complex [((CH3)3tpy)Pt(CCtpy)PtCCCH3]2+ (4′) for DFT calculations.](/image/article/2011/SC/c0sc00427h/c0sc00427h-f8.gif) | ||
| Fig. 8 Model complex [((CH3)3tpy)Pt(CCtpy)PtCCCH3]2+ (4′) for DFT calculations. | ||
In order to have phosphorescence occur after electron excitation, intersystem crossing (ISC) has to be effective to move the complex from an excited singlet state to a triplet excited state via spin–orbit coupling (SOC). Scheme 2 presents the ordering of electronic excited states of 4′ in CH2Cl2 and CH3CN at S0 optimized geometry.
![Excited state orderings of [((CH3)3tpy)Pt(CCtpy)PtCCCH3]2+ at S0 optimized geometry.](/image/article/2011/SC/c0sc00427h/c0sc00427h-s2.gif) | ||
| Scheme 2 Excited state orderings of [((CH3)3tpy)Pt(CCtpy)PtCCCH3]2+ at S0 optimized geometry. | ||
In order to have efficient ISC, the two coupling singlet and triplet excited states have to be close in energy and the d orbitals for the coupling excited states have to be different. In CH2Cl2, T2 is only 173 cm−1 above S1, and thus ISC is effective. However, in CH3CN, T3, which is of the same parentage as T2 in CH2Cl2, is 426 cm−1 above S1 and ISC is less effective compared with that in CH2Cl2. Moreover, there is a low-lying triplet excited state (T2) that is only 511 cm−1 below T3 and internal conversion (IC) from T3 to T2 should be rapid. This T2 excited state, which is derived from the d orbitals of Pt(A) (Pt coordinated to the tpy rings with CH3 substituents), has to couple with the high-lying singlet excited state (> 4500 cm−1) for phosphorescence to occur. Thus, triplet emission quantum yield is low in CH3CN.
In conclusion, complexes 3–6 having tandem two to three {(tpy)Pt} units exhibit interesting aggregation properties that are absent for the monomeric complexes 1 and 2. Among the complexes studied in this work, [(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2 (4) has the highest emission quantum yield (0.43) and the longest lifetime (11 μs) in CH2Cl2 solution. Complex 4 forms “contact ion pairs” in chlorinated solvents with low dielectric constant, but “solvent-separated ion pairs” in CH3CN with high dielectric constant. Based on the results of 1H NMR and emission experiments, we suggest that intermolecular aggregation between cations of 4 would prevail in CH3CN. As the composition of chlorinated solvent in CH3CN increases or in pure chlorinated solvent, the concentration of “contact ion pairs” of complex 4 would increase. Subsequently, suppression of intermolecular aggregation between cations of 4 and restriction of intramolecular motion would enhance the emission intensity at 577 nm by enhancing radiative decay. DFT calculations revealed that the ISC and radiative decay are less effective in CH3CN than in CH2Cl2, thus contributing to low emission quantum yield in CH3CN. The spectacular solvent effect on the luminescent behaviour of complex 4 can be accounted for by the “ion pair” formation, aggregation behaviour, structural conformation and excited state ordering in which all these can be attributed to the planar coordination geometry of platinum(II).
Acknowledgements
We gratefully acknowledge financial support from the Innovation and Technology Commission of the HKSAR Government (ITF/016/08NP), the University Grants Committee of the Hong Kong SAR (AoE/P-03/08), Research Grants Council of Hong Kong SAR (HKU 7008/09P), National Natural Science Foundation of China/Research Grants Council Joint Research Scheme [N_HKU 752/08] and The Chinese Academy of Sciences-Croucher Foundation Funding Scheme For Joint Laboratories. Dr S. C. F. Kui acknowledges the small project funding (200807176030) and the post-doctoral fellow scholarship from The University of Hong Kong.Notes and references
- (a) J. W. Schindler, R. C. Fukuda and A. W. Adamson, J. Am. Chem. Soc., 1982, 104, 3596 CrossRef CAS; (b) V. M. Miskowski, V. H. Houlding, C.-M. Che and Y. Wang, Inorg. Chem., 1993, 32, 2518 CrossRef CAS; (c) H.-K. Yip, H.-M. Lin, Y. Wang and C.-M. Che, Inorg. Chem., 1993, 32, 3402 CrossRef CAS.
- (a) D. M. Roundhill, H. B. Gray and C.-M. Che, Acc. Chem. Res., 1989, 22, 55 CrossRef CAS; (b) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529 CrossRef CAS; (c) V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145 CrossRef CAS; (d) M. G. Hill, J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1996, 35, 4585 CrossRef CAS; (e) B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28 CrossRef CAS.
- (a) K. W. Jennette, J. T. Gill, J. A. Sadownick and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 6159 CrossRef CAS; (b) H.-K. Yip, C.-M. Che, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1992, 1369 RSC; (c) H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933 RSC; (d) J. A. Bally, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1993, 32, 369 CrossRef CAS.
- (a) V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS; (b) V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Chem.–Eur. J., 2002, 8, 4066 CrossRef CAS; (c) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, Angew. Chem., Int. Ed., 2003, 42, 1400 CrossRef; (d) K. M.-C. Wong, W.-S. Tang, B. W.-K. Chu, N. Zhu and V. W.-W. Yam, Organometallics, 2004, 23, 3459 CrossRef CAS.
- (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS; (b) C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791 CrossRef CAS; (c) V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and N. Zhu, Chem.–Eur. J., 2005, 11, 4535 CrossRef.
- (a) T. K. Aldridge, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722 CrossRef CAS; (b) J. F. Michalec, S. A. Bejune and D. R. McMillin, Inorg. Chem., 2000, 39, 2708 CrossRef CAS; (c) D. R. McMillin and J. J. Moore, Coord. Chem. Rev., 2002, 229, 113 CrossRef CAS.
- (a) J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS; (b) R. Büchner, J. S. Field, R. J. Haines, C. T. Cunningham and D. R. McMillin, Inorg. Chem., 1997, 36, 3952 CrossRef; (c) R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711 RSC; (d) T. J. Wadas, Q.-M. Wang, Y.-J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841 CrossRef CAS.
- (a) R. S. Osborn and D. Rogers, J. Chem. Soc., Dalton Trans., 1974, 1002 RSC; (b) W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS; (c) R. H. Herber, M. Croft, M. J. Coyer, B. Bilash and A. Sahiner, Inorg. Chem., 1994, 33, 2422 CrossRef CAS; (d) W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261 CrossRef CAS.
- M.-X. Zhu, W. Lu, N. Zhu and C.-M. Che, Chem.–Eur. J., 2008, 14, 9736 CrossRef CAS.
- V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169 CrossRef CAS.
- (a) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659 CrossRef CAS; (b) W. Lu, Y.-C. Law, J. Han, S. S.-Y. Chui, D.-L. Ma, N. Zhu and C.-M. Che, Chem.–Asian J., 2008, 3, 59 CrossRef CAS; (c) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS.
- M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS.
- (a) S. C. Chan, M. C. W. Chan, Y. Wang, C.-M. Che, K. K. Cheung and N. Zhu, Chem.–Eur. J., 2001, 7, 4180 CrossRef CAS; (b) K. Sonogashira, Y. Fujikura, T. Yatake, N. Toyoshima, S. Takahashi and N. Hagihara, J. Organomet. Chem., 1978, 145, 101 CrossRef CAS.
- (a) B. Manimaran, P. Thanasekaran, T. Rajendran, R.-J. Lin, I.-J. Chang, G.-H. Lee, S.-M. Peng, S. Rajagopal and K.-L. Lu, Inorg. Chem., 2002, 41, 5323 CrossRef CAS; (b) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (c) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS.
- (a) G. Mamantov and A. I. Popov (Eds), Chemistry of Nonaqueous Solutions – Current Progress, VCH, New York, 1994 Search PubMed; (b) J. R. Chipperfield, Non-aqueous Solvents, Oxford University Press, New York, 1999 Search PubMed.
- G. S. M. Tong, Y.-C. Law, S. C. F. Kui, N. Zhu, K. H. Leung, D. L. Phillips and C.-M. Che, Chem.–Eur. J., 2010, 16, 6540 CAS.
- (a) R. Ziessel and S. Diring, Tetrahedron Lett., 2006, 47, 4687 CrossRef CAS; (b) R. Ziessel, S. Diring, P. Kadjane, L. Charbonnière, P. Retailleau and C. Philouze, Chem.–Asian J., 2007, 2, 975 CrossRef CAS; (c) M. L. Muro, S. Diring, X. Wang, R. Ziessel and F. N. Castellano, Inorg. Chem., 2008, 47, 6796 CrossRef CAS.
- V. Grosshenny, F. M. Romero and R. Ziessel, J. Org. Chem., 1997, 62, 1491 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c0sc00427h |
‡ General procedures. The solvents used for synthesis were analytical grade. [(tBu3tpy)PtCCtBu](OTf) (1),16[(tBu3tpy)PtCCtpy](OTf) (2),17 [(COD)PtCl2] (COD = 1,5-cyclooctadiene), HCCtpy (4′-ethynyl-2,2′:6′,2′′-terpyridine)18 were synthesized according to literature methods. Fast atom bombardment (FAB) mass spectra were obtained on a Finnigan Mat 95 mass spectrometer using 3-nitrobenzyl alcohol as matrix. Electrospray Ionization (ESI) mass spectra were obtained on a Finnigan LCQ quadrupole ion trap mass spectrometer (samples were dissolved in HPLC grade acetonitrile). 1H NMR spectra were recorded on a Bruker AVANCE 600, DRX 400 and 500 FT-NMR spectrometers. The 1H NMR chemical shifts were referenced to those of the residual proton/carbon atoms of CD3CN and DMF-d7. Peak assignments were based on 1H–1H, and NOESY 2-D NMR experiments. UV-visible absorption spectra were obtained on a Hewlett-Packard 8453 diode array spectrophotometer. Emission spectra were measured on a Spex Fluorolog-3 spectrofluorometer. Elemental analyses were performed by the Institute of Chemistry at the Chinese Academy of Science, Beijing. TEM were performed on Philips Tecnai G2 20 S-TWIN with an accelerating voltage of 200 kV. The TEM images were taken by a Gatan MultiScan Camera Model 794. The SEM images were taken on a LEO 1530 FEG scanning electron microscope operating at 5.0 kV. TEM samples were prepared by depositing a few drops of MeCN solution or MeCN–Et2O 1:1 solution mixture of the platinum complexes on Formvar® coated copper grids. Excess solvent was removed by a piece of filter paper and this was followed by drying the sample in a vacuum desiccator. SEM samples were prepared by dropping a drop of MeCN solution or MeCN–Et2O 1:1 solution mixture of the platinum complexes onto silicon wafers onto the SEM specimen mounts. All the samples for SEM measurements were sputtered with gold thin film (20 s, < 2 nm thickness).[(tBu3tpy)Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) Ctpy)PtCl](OTf)2(3). [(COD)PtCl2] (5 mg, 0.04 mmol) and 2 (36 mg, 0.036 mmol) were dissolved in a mixture of H2O/acetone (1/4, v/v, 25 mL). The solution was heated at 100 °C for 12 h to give a bright orange solution. Upon removal of solvent, the orange solid was dissolved in warm CH3CN (10 mL) and the solution was filtered into an aqueous solution of NaOTf (34 mg, 2 mL). A reddish orange precipitate was collected on a sintered-glass funnel and air-dried. Yield: 35 mg, 70%. Anal. Calcd for C46H45N6ClO6S2F6Pt2·2H2O: C, 38.97; H, 3.48; N, 5.93. Found: C, 38.90; H, 3.44; N, 5.93. 1H NMR (500 MHz, DMF-d7): δ 1.49 (s, 18H, tBu), 1.56 (s, 9H, tBu), 7.38 (br, 2H, H5′), 7.54 (br, 2H, Hd1), 8.28 (br, s, 2H, He1), 8.41 (t, 2H, JHH = 7.4 Hz, Hc1), 8.48 (br, s, 2H, H6′), 8.61 (s, 2H, H3), 8.70 (d, 2H, JHH = 6.4 Hz, Hb1), 8.80 (s, 2H, H3′), 8.87 (s, 2H, Ha1). FAB-MS (+ve NBA matrix): m/z 1233 [3 − (OTf)]+, 1083 [3 − 2(OTf)]+; ESI-MS (MeCN): m/z 1233 [3 − (OTf)]+, 542 [3 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2097; 2122 cm−1.[(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2(4). Ligand HCCtBu (2.3 mg, 0.028 mmol) was added to a solution of 3 (35 mg, 0.025 mmol), CuI (1 mg, 0.005 mmol), and diisopropylamine (0.5 mL) in CH2Cl2–CH3CN (1/1, v/v, 20 mL). The resultant solution, after stirring for 12 h at room temperature under an argon atmosphere, was evaporated to dryness in vacuo. The crude product was purified by chromatography on an alumina (neutral) column with (CH2Cl2–MeCN) (1/1, v/v) as eluent. Orange needles were obtained by vapor diffusion of diethyl ether into an acetonitrile solution. Yield: 20 mg, 56%. Anal. Calcd for C52H54N6O6S2F6Pt2·2H2O: C, 42.68; H, 4.00; N, 5.74. Found: C, 42.61; H, 3.81; N, 6.10. 1H NMR (500 MHz, CD3CN): δ 1.10 (s, 9H, CCtBu), 1.47 (s, 18H, tBu), 1.58 (s, 9H, tBu), 7.23 (br, s, 2H, Hd1), 7.39 (d, 2H, JHH = 4.2 Hz, H5′), 7.93 (t, 2H, JHH = 7.6 Hz, Hc1), 8.13 (s, 2H, H3), 8.21 (br, s, 4H, H3′ and Hb1), 8.30 (s, 2H, JHH = 5.5 Hz, He1), 8.32 (s, 2H, Ha1), 8.49 (d, 2H, JHH = 7.1 Hz, H6′). FAB-MS (+ve NBA matrix): m/z 1278 [4 − (OTf)]+, 1129 [4 − 2(OTf)]+; ESI-MS (MeCN): m/z 1278 [4 − (OTf)]+, 564 [4 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2093; 2124 cm−1.[(tBu3tpy)Pt(CCtpy)PtCCtpy](OTf)2(5). Ligand HCCtpy (11 mg, 0.039 mmol) was added to a solution of 3 (35 mg, 0.025 mmol), CuI (1 mg, 0.005 mmol), and diisopropylamine (0.5 mL) in CH2Cl2–CH3CN (1/1, v/v, 20 mL). After stirring for 12 h at room temperature under an argon atmosphere, the solution was evaporated to dryness in vacuo, and the solid was dissolved in a minimum amount of acetonitrile. The solution was filtered through Celite and the filtrate was stored at 4 °C for 3 days. The light brown precipitate was washed with CH2Cl2 (5 mL) to afford a brownish orange solid. Yield: 30 mg, 52%. Anal. Calcd for C63H55N9O6S2F6Pt2·2H2O: C, 46.18; H, 3.63; N, 7.81. Found: C, 45.78; H, 3.53; N, 7.86. 1H NMR (500 MHz, 353 K, DMF-d7): δ 1.16 (s, 9H, tBu), 1.51 (s, 18H, tBu), 7.46 (br, s, 2H, Hd2), 7.69–7.72 (m, 4H, H5′ and Hd1), 7.99 (s, 2H, Hc2), 8.30 (br, s, 2H, Ha2), 8.36 (t, 2H, JHH = 7.4 Hz, Hc1), 8.59–8.61 (m, 6H, H3, Ha1 and Hb2), 8.72 (br, s, 6H, H3′, Hb1 and He2), 8.78–8.85 (m, 4H, H6′ and He1). FAB-MS (+ve NBA matrix): m/z 1454 [5 − (OTf)]+, 1304 [5 − 2(OTf)]+; ESI-MS (MeCN): m/z 1454 [5 − (OTf)]+, 652 [5 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2095; 2124 cm−1.[(tBu3tpy)Pt(CCtpy)Pt(CCtpy)PtCl](OTf)3(6): [(COD)PtCl2] (15 mg, 0.04 mmol) and 6 (50 mg, 0.030 mmol) were dissolved in H2O/acetone (1/4, v/v, 25 mL). The solution was heated at 100 °C for 24 h to give a red solution, which was filtered into an aqueous solution of NaOTf (50 mg, 2 mL). The brownish orange precipitate was collected and air-dried. Yield: 32 mg, 52%. Anal. Calcd for C64H55N9ClO9S3F9Pt3: C, 38.78; H, 2.80; N, 6.36. Found: C, 38.74; H, 3.10; N, 6.20. 1H NMR (500 MHz, 353 K, DMF-d7): δ 1.67 (s, 18H, tBu), 1.71 (s, 9H, tBu), 7.97–7.99 (br, 2H, H5′), 8.09–8.11 (br, fused with DMF-d7 peak, 2H, Hd2), 8.18–8.21 (br, fused with DMF-d7 peak, 2H, Hd1), 8.66 (t, J = 8.0 Hz, 2H, Hc2), 8.86–8.88 (m, 4H, H3 and Hb2), 8.90–8.95 (m, 8 H, H3′, Ha1, Hb1 and Hc1), 9.01 (s, 2H, Ha2), 9.06 (m, 2H, He2), 9.14 (d, J = 6.1 Hz, 2H, He1), 9.24 (m, 2H, Hb1). FAB-MS (+ve NBA matrix): m/z 1834 [6 − (OTf)]+, 1685 [6 − 2(OTf)]+, 1534 [6 − 3(OTf)]+; ESI-MS (MeCN): m/z 1833 [6 − (OTf)]+. IR (Nujol): vmax (CC) 2097; 2124 cm−1. Ctpy)PtCl](OTf)2(3). [(COD)PtCl2] (5 mg, 0.04 mmol) and 2 (36 mg, 0.036 mmol) were dissolved in a mixture of H2O/acetone (1/4, v/v, 25 mL). The solution was heated at 100 °C for 12 h to give a bright orange solution. Upon removal of solvent, the orange solid was dissolved in warm CH3CN (10 mL) and the solution was filtered into an aqueous solution of NaOTf (34 mg, 2 mL). A reddish orange precipitate was collected on a sintered-glass funnel and air-dried. Yield: 35 mg, 70%. Anal. Calcd for C46H45N6ClO6S2F6Pt2·2H2O: C, 38.97; H, 3.48; N, 5.93. Found: C, 38.90; H, 3.44; N, 5.93. 1H NMR (500 MHz, DMF-d7): δ 1.49 (s, 18H, tBu), 1.56 (s, 9H, tBu), 7.38 (br, 2H, H5′), 7.54 (br, 2H, Hd1), 8.28 (br, s, 2H, He1), 8.41 (t, 2H, JHH = 7.4 Hz, Hc1), 8.48 (br, s, 2H, H6′), 8.61 (s, 2H, H3), 8.70 (d, 2H, JHH = 6.4 Hz, Hb1), 8.80 (s, 2H, H3′), 8.87 (s, 2H, Ha1). FAB-MS (+ve NBA matrix): m/z 1233 [3 − (OTf)]+, 1083 [3 − 2(OTf)]+; ESI-MS (MeCN): m/z 1233 [3 − (OTf)]+, 542 [3 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2097; 2122 cm−1.[(tBu3tpy)Pt(CCtpy)PtCCtBu](OTf)2(4). Ligand HCCtBu (2.3 mg, 0.028 mmol) was added to a solution of 3 (35 mg, 0.025 mmol), CuI (1 mg, 0.005 mmol), and diisopropylamine (0.5 mL) in CH2Cl2–CH3CN (1/1, v/v, 20 mL). The resultant solution, after stirring for 12 h at room temperature under an argon atmosphere, was evaporated to dryness in vacuo. The crude product was purified by chromatography on an alumina (neutral) column with (CH2Cl2–MeCN) (1/1, v/v) as eluent. Orange needles were obtained by vapor diffusion of diethyl ether into an acetonitrile solution. Yield: 20 mg, 56%. Anal. Calcd for C52H54N6O6S2F6Pt2·2H2O: C, 42.68; H, 4.00; N, 5.74. Found: C, 42.61; H, 3.81; N, 6.10. 1H NMR (500 MHz, CD3CN): δ 1.10 (s, 9H, CCtBu), 1.47 (s, 18H, tBu), 1.58 (s, 9H, tBu), 7.23 (br, s, 2H, Hd1), 7.39 (d, 2H, JHH = 4.2 Hz, H5′), 7.93 (t, 2H, JHH = 7.6 Hz, Hc1), 8.13 (s, 2H, H3), 8.21 (br, s, 4H, H3′ and Hb1), 8.30 (s, 2H, JHH = 5.5 Hz, He1), 8.32 (s, 2H, Ha1), 8.49 (d, 2H, JHH = 7.1 Hz, H6′). FAB-MS (+ve NBA matrix): m/z 1278 [4 − (OTf)]+, 1129 [4 − 2(OTf)]+; ESI-MS (MeCN): m/z 1278 [4 − (OTf)]+, 564 [4 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2093; 2124 cm−1.[(tBu3tpy)Pt(CCtpy)PtCCtpy](OTf)2(5). Ligand HCCtpy (11 mg, 0.039 mmol) was added to a solution of 3 (35 mg, 0.025 mmol), CuI (1 mg, 0.005 mmol), and diisopropylamine (0.5 mL) in CH2Cl2–CH3CN (1/1, v/v, 20 mL). After stirring for 12 h at room temperature under an argon atmosphere, the solution was evaporated to dryness in vacuo, and the solid was dissolved in a minimum amount of acetonitrile. The solution was filtered through Celite and the filtrate was stored at 4 °C for 3 days. The light brown precipitate was washed with CH2Cl2 (5 mL) to afford a brownish orange solid. Yield: 30 mg, 52%. Anal. Calcd for C63H55N9O6S2F6Pt2·2H2O: C, 46.18; H, 3.63; N, 7.81. Found: C, 45.78; H, 3.53; N, 7.86. 1H NMR (500 MHz, 353 K, DMF-d7): δ 1.16 (s, 9H, tBu), 1.51 (s, 18H, tBu), 7.46 (br, s, 2H, Hd2), 7.69–7.72 (m, 4H, H5′ and Hd1), 7.99 (s, 2H, Hc2), 8.30 (br, s, 2H, Ha2), 8.36 (t, 2H, JHH = 7.4 Hz, Hc1), 8.59–8.61 (m, 6H, H3, Ha1 and Hb2), 8.72 (br, s, 6H, H3′, Hb1 and He2), 8.78–8.85 (m, 4H, H6′ and He1). FAB-MS (+ve NBA matrix): m/z 1454 [5 − (OTf)]+, 1304 [5 − 2(OTf)]+; ESI-MS (MeCN): m/z 1454 [5 − (OTf)]+, 652 [5 − 2(OTf)]2+. IR (Nujol): vmax (CC) 2095; 2124 cm−1.[(tBu3tpy)Pt(CCtpy)Pt(CCtpy)PtCl](OTf)3(6): [(COD)PtCl2] (15 mg, 0.04 mmol) and 6 (50 mg, 0.030 mmol) were dissolved in H2O/acetone (1/4, v/v, 25 mL). The solution was heated at 100 °C for 24 h to give a red solution, which was filtered into an aqueous solution of NaOTf (50 mg, 2 mL). The brownish orange precipitate was collected and air-dried. Yield: 32 mg, 52%. Anal. Calcd for C64H55N9ClO9S3F9Pt3: C, 38.78; H, 2.80; N, 6.36. Found: C, 38.74; H, 3.10; N, 6.20. 1H NMR (500 MHz, 353 K, DMF-d7): δ 1.67 (s, 18H, tBu), 1.71 (s, 9H, tBu), 7.97–7.99 (br, 2H, H5′), 8.09–8.11 (br, fused with DMF-d7 peak, 2H, Hd2), 8.18–8.21 (br, fused with DMF-d7 peak, 2H, Hd1), 8.66 (t, J = 8.0 Hz, 2H, Hc2), 8.86–8.88 (m, 4H, H3 and Hb2), 8.90–8.95 (m, 8 H, H3′, Ha1, Hb1 and Hc1), 9.01 (s, 2H, Ha2), 9.06 (m, 2H, He2), 9.14 (d, J = 6.1 Hz, 2H, He1), 9.24 (m, 2H, Hb1). FAB-MS (+ve NBA matrix): m/z 1834 [6 − (OTf)]+, 1685 [6 − 2(OTf)]+, 1534 [6 − 3(OTf)]+; ESI-MS (MeCN): m/z 1833 [6 − (OTf)]+. IR (Nujol): vmax (CC) 2097; 2124 cm−1. |
| This journal is © The Royal Society of Chemistry 2011 |